ELISA vs PCR for Allergen Detection in Processed Foods: A Scientific Comparison for Researchers
This article provides a comprehensive comparison of ELISA (Enzyme-Linked Immunosorbent Assay) and PCR (Polymerase Chain Reaction) methodologies for allergen detection in processed food matrices.
ELISA vs PCR for Allergen Detection in Processed Foods: A Scientific Comparison for Researchers
Abstract
This article provides a comprehensive comparison of ELISA (Enzyme-Linked Immunosorbent Assay) and PCR (Polymerase Chain Reaction) methodologies for allergen detection in processed food matrices. Tailored for researchers, scientists, and drug development professionals, it explores the foundational principles, optimal applications, and limitations of each technique. The content covers methodological considerations for complex and processed foods, troubleshooting for common challenges, and validation strategies through comparative data. By synthesizing current research and technological advances, this review serves as a strategic guide for selecting and optimizing allergen detection methods to ensure food safety and regulatory compliance.
Core Principles: Understanding ELISA and PCR Technologies in Allergen Science
This guide provides an in-depth examination of the enzyme-linked immunosorbent assay (ELISA) fundamental mechanism, focusing on its core principle of antigen-antibody interaction for protein detection. Within the broader context of analytical techniques for food allergen detection, we objectively compare ELISA's performance with polymerase chain reaction (PCR)-based methods, presenting experimental data on sensitivity, specificity, and applicability across various food matrices. For researchers and drug development professionals, this analysis delineates the specific technological niches where each method excels, supported by recent comparative studies and standardized protocols.
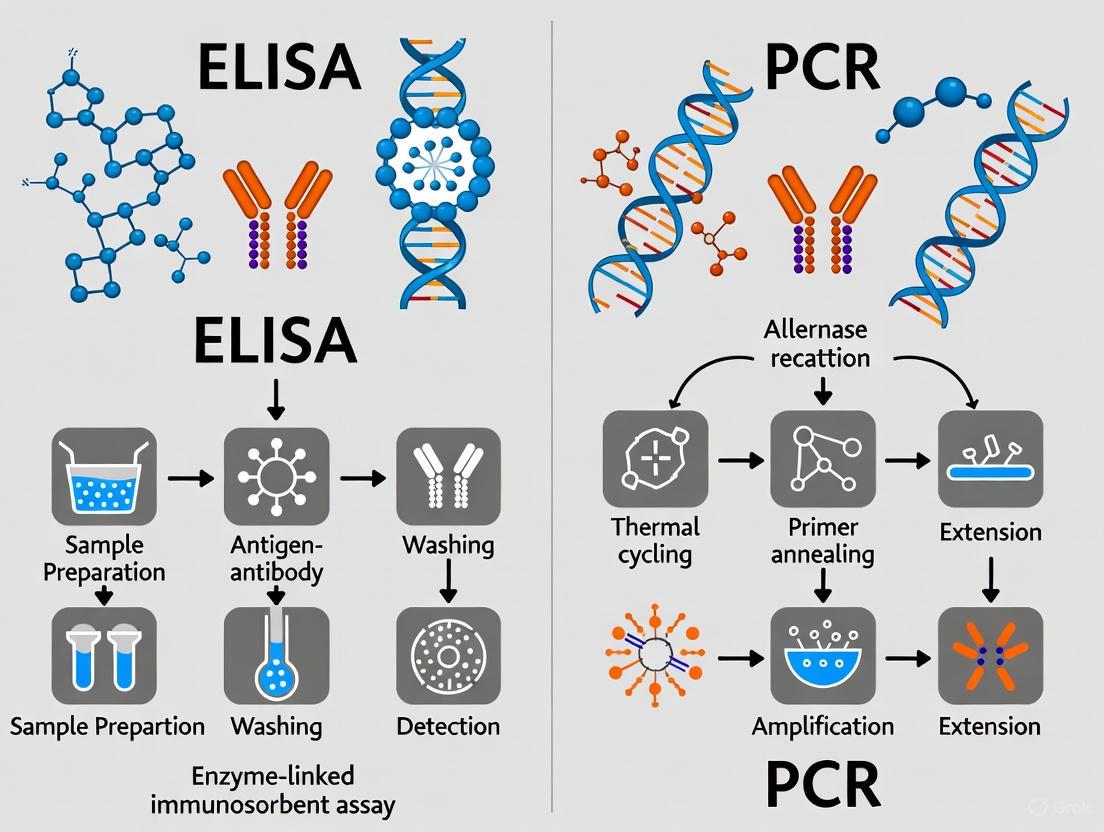
The enzyme-linked immunosorbent assay (ELISA) is a plate-based technique designed for detecting and quantifying soluble substances such as peptides, proteins, antibodies, and hormones [1]. The method was originally described by Engvall and Perlmann in 1971 and relies on the highly specific interaction between an antibody and its target antigen to analyze protein samples immobilized in microplate wells [1]. This interaction forms the foundational basis for ELISA's application across diverse fields, including clinical diagnostics, therapeutic development, and food safety analysis.
In food allergen detection, the accurate identification and quantification of specific allergenic proteins is crucial for public health protection. Food allergies constitute a significant global health concern, and since avoidance is the primary preventive measure, sensitive and accurate detection methods are essential [2] [3]. ELISA has emerged as a cornerstone technology in this domain, offering the sensitivity and specificity required for regulatory compliance and quality control [4] [5]. The Codex Alimentarius Commission has formally adopted ELISA as the official test for gluten allergens, specifying that gluten levels in food must not exceed 20 mg/kg [3].
Fundamental Mechanism of ELISA
The fundamental mechanism of ELISA centers on the specific binding between an antigen and an antibody, with the detection achieved through an enzyme-mediated colorimetric reaction. All ELISA variants share a common fundamental workflow consisting of four core steps: coating/capture, plate blocking, probing/detection, and signal measurement [1]. The critical element ensuring assay specificity is the antibody-antigen interaction, while sensitivity is achieved through enzyme-mediated signal amplification.
Core Steps in ELISA Protocol
The ELISA procedure follows a sequential process that ensures specific antigen capture and sensitive detection:
- Coating/Capture: Direct or indirect immobilization of antigens to the surface of polystyrene microplate wells [1]. This is typically achieved through passive adsorption via hydrophobic interactions between the plastic and non-polar protein residues [1].
- Plate Blocking: Addition of an irrelevant protein (e.g., bovine serum albumin) or other molecule to cover all unsaturated surface-binding sites of the microplate wells, thereby reducing non-specific binding [6] [7].
- Probing/Detection: Incubation with antigen-specific antibodies that affinity-bind to the target antigens [1]. This may involve primary and secondary antibody systems depending on the ELISA format.
- Signal Measurement: Detection of the signal generated via an enzyme-linked conjugate and its substrate [1]. Common enzyme labels include horseradish peroxidase (HRP) and alkaline phosphatase (AP), with substrates selected for colorimetric, fluorescent, or chemiluminescent detection [1].
Key ELISA Formats and Their Mechanisms
Among several ELISA formats, the sandwich ELISA is predominant for allergen detection due to its high specificity and sensitivity [4]. The diagram below illustrates the fundamental mechanism and procedural workflow of a sandwich ELISA.
Diagram 1: ELISA Mechanism and Workflow. This illustrates the sequential steps of a sandwich ELISA and the molecular interactions at each stage, culminating in signal generation.
The sandwich ELISA format, which "sandwiches" the target antigen between two specific antibodies, provides exceptional specificity because it requires simultaneous recognition by two different antibodies [1] [4]. This dual-antibody recognition minimizes false positives, making it particularly valuable for detecting specific allergenic proteins in complex food matrices [4]. For low-molecular-weight targets that cannot accommodate two antibodies, competitive ELISA formats are employed, where the antigen in the sample competes with a labeled reference for binding to a limited amount of antibody [1] [6].
Comparative Performance: ELISA vs. PCR in Food Allergen Detection
When selecting an appropriate method for allergen detection in processed foods, researchers must consider the fundamental differences in what each technique detects: ELISA targets specific allergenic proteins, while PCR detects species-specific DNA sequences. The table below summarizes the core characteristics of each method.
Table 1: Fundamental Comparison of ELISA and PCR Methodologies
| Parameter | ELISA (Enzyme-Linked Immunosorbent Assay) | PCR (Polymerase Chain Reaction) |
|---|---|---|
| Detection Target | Allergenic proteins (the actual allergens) [4] [5] | DNA from allergenic ingredients [4] [5] |
| Principle | Antigen-antibody interaction and enzyme-mediated color development [1] [6] | Amplification of species-specific DNA sequences [4] |
| Sensitivity | High (sensitive to low protein levels) [4] | Very high (detects trace DNA) [4] |
| Quantification | Quantitative (measures allergen concentration) [4] [5] | Generally qualitative or semi-quantitative [4] |
| Time to Result | ~2-3 days [4] | ~4-5 days [4] |
Experimental Data from Comparative Studies
Direct comparative studies reveal how these fundamental differences translate into practical performance for detecting allergens in various food matrices. The following table compiles experimental findings from recent research.
Table 2: Experimental Performance Data for Allergen Detection
| Study Focus & Matrix | Method | Key Performance Findings | Source |
|---|---|---|---|
| Crustacean Allergens (Manhattan clam chowder, fish sauce) | Real-time PCR | Broader dynamic range (0.1–10⁶ mg/kg); no significant matrix interference observed. | [2] |
| ELISA | Narrower dynamic range (200–4000 mg/kg); exhibited matrix interference. | [2] | |
| Beef & Pork Detection (Processed meat products) | Real-time PCR | Detected pork at 0.10% and beef at 0.50% in binary mixtures. | [8] |
| ELISA | Detected pork at 10.0% and beef at 1.00% in binary mixtures. | [8] | |
| General Allergen Detection (Various foods) | PCR | Preferred for highly processed foods, complex matrices, and when target is celery/fish. More stable DNA survives heat/pressure/pH changes. | [4] [5] |
| ELISA | Standard method for gluten; preferred for egg, milk, and when quantitative results are required. Proteins may degrade with processing. | [4] [5] |
The data indicate that real-time PCR often demonstrates higher sensitivity and a broader dynamic range in complex matrices, as it is less susceptible to matrix interference than ELISA [2] [8]. However, a significant limitation of PCR is its indirect nature; it detects DNA, not the allergenic protein itself, which can lead to false positives if the DNA is present from a non-allergenic source or if the allergenic protein has been removed [3]. Conversely, ELISA's direct measurement of the protein allergen can be compromised by food processing that denatures the protein's structure, altering antibody-binding epitopes and potentially leading to false negatives [3].
Detailed Experimental Protocols
To ensure reproducibility and reliability in research applications, standardized protocols for both ELISA and PCR are essential. The following sections detail the core methodologies.
Detailed Protocol: Sandwich ELISA for Allergen Detection
The following protocol is adapted from established methodologies for food allergen analysis [7]:
- Plate Coating: Dilute the capture antibody in phosphate-buffered saline (PBS, pH 7.4) or carbonate-bicarbonate buffer (pH 9.4) to a concentration of 2–10 μg/mL. Add 100 μL per well to a 96-well polystyrene microplate. Seal the plate to prevent evaporation and incubate at 4°C for 15-18 hours to immobilize the antibody via hydrophobic interactions [1] [7].
- Washing: Remove the coating solution. Wash the plate three times with washing solution (e.g., PBS containing 0.05% Tween 20) to remove unbound antibody [7].
- Blocking: Add 200-300 μL of blocking buffer (e.g., 1% BSA or 5% non-fat dry milk in PBS) to each well. Incubate at 37°C for 1 hour to cover any remaining protein-binding sites [7].
- Washing: Remove the blocking buffer and wash the plate three times with washing solution [7].
- Sample Addition: Dilute the food sample extract in an appropriate sample dilution buffer. Add 100 μL of the diluted sample or antigen standard (for the calibration curve) to each well. Incubate at 37°C for 1 hour to allow the target antigen to bind to the capture antibody [7].
- Washing: Remove the sample and wash the plate five times with washing solution to remove unbound material [7].
- Detection Antibody Incubation: Dilute the specific, biotinylated or enzyme-labeled detection antibody in sample dilution buffer. Add 100 μL to each well and incubate at 37°C for 1 hour. This antibody must recognize a different epitope on the target antigen than the capture antibody [7].
- Washing: Remove the detection antibody and wash the plate five times with washing solution [7].
- Enzyme Conjugate Incubation (if needed): For indirect detection, add an enzyme-conjugated secondary antibody (e.g., HRP-labeled anti-species IgG) and incubate for 1 hour at 37°C, followed by another five washes [7].
- Signal Development: Add 100 μL of enzyme substrate solution (e.g., TMB for HRP) to each well. Incubate at room temperature in the dark for 15-30 minutes to allow color development [6] [7].
- Reaction Stopping: Add 100 μL of stop solution (e.g., 1M sulfuric acid for TMB) to each well to terminate the enzyme reaction [7].
- Absorbance Measurement: Immediately measure the absorbance of each well at the appropriate wavelength (e.g., 450 nm for TMB) using a microplate reader. The antigen concentration in the sample is determined by interpolation from the standard curve [6] [7].
While a full protocol is beyond this guide's scope, the core workflow for PCR-based allergen detection includes [4]:
- DNA Extraction: Isolate total DNA from the food sample using a commercial kit suitable for the specific matrix.
- Primer/Probe Design: Design oligonucleotide primers and probes (e.g., TaqMan) that target a species-specific DNA sequence (e.g., mitochondrial 12S rRNA, cytochrome b gene) [2] [8].
- Amplification Reaction: Prepare a reaction mix containing the extracted DNA, primers, probes, nucleotides, and a DNA polymerase with hot-start capability. Run the reaction in a real-time PCR instrument with a defined thermal cycling profile (e.g., initial denaturation at 95°C, followed by 40 cycles of denaturation, annealing, and extension) [8].
- Data Analysis: Determine the cycle threshold (Ct) value. The presence of the target allergen is confirmed by a Ct value below a validated cutoff. Quantification, if applied, is achieved by comparison to a standard curve from serial dilutions of known target DNA [4].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of ELISA requires specific, high-quality reagents and equipment. The following table details essential components for establishing a robust sandwich ELISA.
Table 3: Essential Research Reagents and Materials for Sandwich ELISA
| Item | Function/Description | Key Considerations |
|---|---|---|
| Microplate | 96-well or 384-well polystyrene plate for immobilizing biomolecules [1]. | Choose clear for colorimetric, black/white for fluorescent/chemiluminescent signals; ensure high protein-binding capacity and low well-to-well variation [1]. |
| Capture Antibody | The primary antibody immobilized on the plate to specifically bind the target antigen [1]. | Must be highly specific and recognize a different epitope than the detection antibody; often a monoclonal antibody for consistency. |
| Blocking Buffer | A solution of irrelevant protein (e.g., BSA, casein) used to cover unsaturated binding sites [6]. | Prevents non-specific binding of other assay components to the plate; critical for reducing background noise. |
| Detection Antibody | The antibody that binds to a different epitope of the captured antigen [1]. | Often conjugated directly to an enzyme (e.g., HRP) or a tag (e.g., biotin) for subsequent detection. |
| Enzyme-Conjugate | An enzyme-linked secondary antibody or streptavidin used for signal generation [1]. | Must be specific to the detection antibody; common enzymes are Horseradish Peroxidase (HRP) and Alkaline Phosphatase (AP) [6]. |
| Substrate | The molecule converted by the enzyme to a detectable product [1]. | Choice depends on the enzyme and detection method (colorimetric, chemiluminescent, fluorescent); TMB is a common colorimetric substrate for HRP [6]. |
| Wash Buffer | Typically a buffered solution with a mild detergent (e.g., PBS with 0.05% Tween 20) [6]. | Used to remove unbound reagents between steps; critical for achieving a high signal-to-noise ratio. |
| Microplate Reader | Instrument to measure the absorbance, fluorescence, or luminescence of the plate wells [1]. | Must be compatible with the plate format and the chosen detection method (e.g., spectrophotometer for colorimetric signals) [1]. |
The fundamental mechanism of ELISA, rooted in specific antigen-antibody interaction and enzymatic signal amplification, makes it a powerful tool for the direct quantification of protein allergens. Its strengths are particularly evident in quantitative analysis of specific proteins like gluten and in matrices where protein integrity is maintained. However, as the comparative data show, PCR provides a complementary and sometimes superior approach for detecting allergens in highly processed foods or complex ingredients, owing to the stability of DNA. The choice between ELISA and PCR is not a matter of which technology is universally better, but which is more fit-for-purpose given the specific sample matrix, the target molecule, and the required information (qualitative vs. quantitative). For a comprehensive food allergen control program, leveraging the strengths of both methods often provides the most robust strategy for ensuring product safety and regulatory compliance.
The Polymerase Chain Reaction (PCR) is a foundational technique in molecular biology that allows for the exponential amplification of specific DNA sequences in vitro [9]. Since its introduction by Kary Mullis in 1985, for which he was later awarded the Nobel Prize in Chemistry, PCR has revolutionized genetic analysis and become an indispensable tool in biomedical research, clinical diagnostics, and food safety testing [9] [10]. In the specific context of allergen detection in processed foods, PCR offers a powerful DNA-based method to identify the presence of allergenic ingredients, such as crustacean shellfish, by targeting and amplifying species-specific genetic markers, even in complex food matrices [2] [5]. This technique is particularly valuable for verifying allergen-free claims and detecting allergens in processed foods where protein structures may be altered, as DNA is often more stable than proteins through various food processing conditions [5].
This guide will objectively compare the performance of PCR with the protein-based Enzyme-Linked Immunosorbent Assay (ELISA) for allergen detection, providing supporting experimental data to highlight the strengths, limitations, and optimal applications of each technology.
Fundamental Mechanism of PCR
The core principle of PCR is the enzymatic amplification of a specific DNA fragment, defined by two short oligonucleotide primers, through repeated cycles of heating and cooling. The process relies on a thermostable DNA polymerase, typically Taq polymerase isolated from the thermophilic bacterium Thermus aquaticus, which remains active despite repeated exposure to high temperatures [9] [11].
The PCR Cycle
A standard PCR amplification involves three fundamental steps per cycle, repeated 30-40 times [9] [12]:
- Denaturation: The double-stranded DNA template is heated to 94–95°C for 15 seconds to 2 minutes. This high temperature disrupts the hydrogen bonds between complementary base pairs, resulting in two single-stranded DNA molecules [9] [12].
- Annealing: The reaction temperature is lowered to 40–60°C (typically 55–72°C) for 15–60 seconds. This allows the forward and reverse primers to bind (anneal) to their complementary sequences on the single-stranded DNA templates, providing a starting point for DNA synthesis [9] [12].
- Extension: The temperature is raised to the optimum for the DNA polymerase, usually 70–74°C, for 1–2 minutes. Taq polymerase synthesizes a new DNA strand by adding deoxynucleotide triphosphates (dNTPs) to the 3' end of the primers, elongating the DNA sequence complementary to the template strand [9] [12].
Each cycle theoretically doubles the amount of the target DNA fragment, or amplicon, leading to an exponential amplification that can generate billions of copies from a single target sequence in a matter of hours [10] [12].
Key Variations: Real-Time PCR and RT-PCR
For allergen detection, two advanced forms of PCR are particularly relevant:
- Real-Time PCR (qPCR): This method allows for the simultaneous amplification and quantification of target DNA. It uses fluorescent dyes or probes that emit signals proportional to the amount of amplified DNA, enabling real-time monitoring of the reaction. This eliminates the need for post-PCR processing and provides quantitative data [9] [5].
- Reverse Transcription PCR (RT-PCR): When the target is RNA, RT-PCR is used. It first employs a reverse transcriptase enzyme to convert RNA into complementary DNA (cDNA), which is then amplified by standard PCR. This was the primary method for detecting SARS-CoV-2 RNA during the COVID-19 pandemic [9] [12].
Comparative Performance Data: PCR vs. ELISA
The choice between PCR and ELISA for allergen detection depends on the specific application, as each method has distinct performance characteristics. The table below summarizes key comparative data from experimental studies.
Table 1: Performance Comparison of PCR and ELISA for Allergen Detection
| Parameter | PCR (DNA-Based) | ELISA (Protein-Based) | Experimental Context & Data Source |
|---|---|---|---|
| Target Molecule | Species-specific DNA sequences [5] | Allergenic proteins (e.g., tropomyosin) [5] [13] | Fundamental difference in detection principle [5]. |
| Dynamic Range | 0.1 – 106 mg/kg [2] | 200 – 4000 mg/kg [2] | Comparison for crustacean shellfish allergens in food matrices [2]. |
| Sensitivity | High (can detect trace DNA) [5] | High (can detect trace proteins) [6] [13] | Both are highly sensitive, but to different molecular targets [5]. |
| Matrix Interference | Less susceptible in studied models [2] | Observed in complex matrices (e.g., chowder, fish sauce) [2] | Side-by-side comparison in Manhattan clam chowder and fish sauce [2]. |
| Quantification | Preferred for qualitative analysis; qPCR enables quantification [5] | Gold standard for quantitative protein analysis [6] [5] | ELISA is the standard for gluten quantification per Codex Alimentarius [5]. |
| Effect of Food Processing | More reliable for highly processed foods (DNA is stable) [5] | Protein denaturation in processing can affect detection [5] | PCR is preferred for hydrolyzed and fermented samples [5]. |
| Specific Allergen Suitability | Celery, fish, and for species differentiation [5] | Egg, milk, gluten; targets the allergenic protein directly [5] | Celery is low-protein; no common fish antigen for ELISA [5]. |
Experimental Protocols for Allergen Detection
To ensure reproducibility in research, detailed methodologies for both PCR and ELISA are provided below.
Protocol for Real-Time PCR Detection of Allergens
This protocol is adapted from methodologies used for detecting crustacean shellfish allergens [2] [5].
- DNA Extraction: Extract genomic DNA from the food sample using a commercial kit suitable for the specific matrix (e.g., baked goods, sauces). The quality and purity of the DNA are critical for amplification efficiency [5].
- Primer Design: Select primers that target a species-specific gene sequence. Common targets include mitochondrial genes (e.g., 12S rRNA for shrimp, crab, lobster) or the nuclear tropomyosin gene [2] [5].
- Reaction Setup: Prepare the qPCR reaction mix containing:
- Thermal Cycling: Run the reaction in a real-time thermal cycler with a program such as:
- Data Analysis: Determine the quantification cycle (Cq). The presence of the target allergen is confirmed by a Cq value below a predetermined threshold. A standard curve can be used for relative quantification [9].
Protocol for Sandwich ELISA Detection of Allergens
This protocol, based on standard sandwich ELISA procedures, is used for quantifying specific allergenic proteins like tropomyosin [6] [5].
- Coating: Coat a 96-well microplate with a capture antibody specific to the target allergen (e.g., anti-tropomyosin). Incubate overnight at 4°C [6].
- Blocking: Wash the plate with PBS-T (phosphate-buffered saline with Tween-20) to remove unbound antibody. Block remaining protein-binding sites with a blocking agent like bovine serum albumin (BSA) or synthetic block buffer for 1-2 hours at room temperature [6].
- Sample Incubation: Prepare the food sample by extracting proteins in a suitable buffer, often at high temperatures. Add the sample extract or standard to the wells and incubate for 1-2 hours at 37°C. The target allergen binds to the capture antibody [5] [13].
- Detection Antibody Incubation: Wash the plate and add an enzyme-linked detection antibody (conjugated with HRP or AP) specific to a different epitope on the allergen. Incubate for 1-2 hours at room temperature [6].
- Signal Development and Measurement: Wash the plate to remove unbound detection antibody. Add an enzyme substrate (e.g., TMB for HRP). The enzyme catalyzes a reaction that produces a color change. Stop the reaction with an acid (e.g., sulfuric acid) and measure the absorbance of the solution with a spectrophotometer [6] [14]. The color intensity is proportional to the concentration of the allergen in the sample.
Decision Workflow: Selecting the Appropriate Method
The following diagram outlines a logical workflow to guide researchers in selecting the most appropriate method based on their experimental goal and sample characteristics.
The Scientist's Toolkit: Essential Research Reagents
Successful implementation of PCR and ELISA requires specific, high-quality reagents. The following table details the essential components for each method.
Table 2: Essential Research Reagents for PCR and ELISA
| Method | Reagent/Material | Function | Key Considerations |
|---|---|---|---|
| PCR | Taq DNA Polymerase | Thermostable enzyme that synthesizes new DNA strands [9] [10]. | Thermostability is critical for repeated heating cycles. |
| Primers | Short, single-stranded DNA sequences that define the start and end of the target DNA region to be amplified [9]. | Must be specific to the target allergen's DNA sequence; design is critical for specificity. | |
| dNTPs | Deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP); the building blocks for new DNA strands [12]. | Quality and concentration affect amplification efficiency and fidelity. | |
| Thermal Cycler | Instrument that automates the temperature cycles required for PCR [9] [10]. | Must provide precise temperature control and rapid transitions. | |
| Fluorescent Probes/Dyes | Molecules that fluoresce when bound to double-stranded DNA, enabling real-time detection in qPCR [9]. | Allows for quantification of the amplified product. | |
| ELISA | Microplate | 96-well polystyrene plate that serves as the solid phase for the immunoassay [6]. | Must have high protein-binding capacity. |
| Capture & Detection Antibodies | Antibodies that bind specifically to the target allergenic protein [6] [13]. | Specificity is paramount; "matched pairs" are needed for sandwich ELISA. | |
| Enzyme Conjugate | Detection antibody linked to an enzyme (e.g., Horseradish Peroxidase - HRP). | Catalyzes the signal-generating reaction [6]. | |
| Enzyme Substrate | Compound (e.g., TMB) converted by the enzyme to a colored product [6]. | Signal intensity is proportional to allergen concentration. | |
| Blocking Buffer | (e.g., BSA) used to coat unused protein-binding sites on the microplate. | Reduces nonspecific binding, minimizing background noise [6]. |
Both PCR and ELISA are powerful techniques for allergen detection in processed foods, yet they operate on fundamentally different principles. PCR excels in sensitivity and specificity for DNA-based identification, proving particularly robust for detecting allergens in highly processed foods and for species differentiation. Conversely, ELISA directly quantifies the allergenic protein itself and remains the gold standard for compliance testing where regulatory thresholds are defined in protein concentration.
The choice between these methods is not a matter of superiority but of context. Researchers and food safety professionals must base their selection on the specific experimental or monitoring goal, considering the nature of the allergen, the complexity of the food matrix, and the required output (qualitative vs. quantitative). As demonstrated in comparative studies, an integrated approach using both methods can often provide the most comprehensive and reliable assurance of food safety, leveraging the complementary strengths of DNA and protein analysis to protect consumers effectively.
In the field of food allergen detection, two methodologies dominate research and routine analysis: the enzyme-linked immunosorbent assay (ELISA) and the polymerase chain reaction (PCR). These techniques are fundamentally distinct in their analytical targets. ELISA is a protein-based immunochemical method that directly detects the allergenic proteins themselves, which are the actual molecules that trigger immune responses in sensitized individuals [15] [5]. In contrast, PCR is a DNA-based molecular biology technique that detects specific DNA sequences unique to the allergenic source (e.g., peanut, crustacean, wheat) [16] [4]. This core distinction—direct protein detection versus indirect genetic marker detection—dictates their respective performance characteristics, advantages, and limitations, particularly when applied to processed food matrices where both proteins and DNA can undergo significant structural alteration.
The choice between ELISA and PCR is not merely a matter of preference but a strategic decision based on the research question, the nature of the food matrix, and the processing history of the sample. This guide provides an objective, data-driven comparison of these two cornerstone technologies to inform researchers, scientists, and drug development professionals in selecting the optimal method for their specific application.
Performance Comparison: Sensitivity, Specificity, and Limitations
Extensive comparative studies have quantified the performance differences between ELISA and PCR across various food allergens and matrices. The following table summarizes key experimental findings from direct comparison studies.
Table 1: Experimental Performance Data from Direct Method Comparisons
| Study Focus | Method | Reported Sensitivity (LOD/LOQ) | Dynamic Range | Key Finding | Reference |
|---|---|---|---|---|---|
| Crustacean Shellfish (Shrimp, Crab, Lobster) | Real-time PCR | Broader dynamic range | 0.1 - 100,000 mg/kg | No significant matrix interference observed in tested matrices. | [2] |
| ELISA (Total Crustacean) | More limited dynamic range | 200 - 4,000 mg/kg | Showed matrix interference in some samples. | [2] | |
| Beef in Meat Products | Real-time PCR | Consistent detection at 0.50% (w/w) | N/R | Greater sensitivity and agreement among duplicates. | [8] |
| ELISA | Consistent detection at 1.00% (w/w) | N/R | Less time-consuming and easier to perform. | [8] | |
| Pork in Meat Products | Real-time PCR | Consistent detection at 0.10% (w/w) | N/R | Optimal for low detection limits in processed products. | [8] |
| ELISA | Consistent detection at 10.0% (w/w) | N/R | 100% specificity, but significantly less sensitive for pork. | [8] | |
| Peanut in Processed Foods | Real-time PCR | <10 ppm | N/R | Detected one more positive sample than ELISA in market survey. | [17] |
| Sandwich ELISA | <10 ppm | N/R | Results correlated well with PCR; a reliable tool for hidden allergens. | [17] |
Abbreviations: LOD (Limit of Detection), LOQ (Limit of Quantification), N/R (Not Reported in the context of the study).
A critical limitation of ELISA is its susceptibility to producing false-negative results in processed foods. Heat treatment, fermentation, and hydrolysis can denature proteins, altering the conformational epitopes recognized by the antibodies used in ELISA kits [18]. If the antibody cannot bind to the denatured protein, the allergen will not be detected, even if it retains its allergenic potential [15]. PCR can circumvent this issue, as DNA is generally more stable than proteins under such conditions [16] [5]. However, a positive PCR signal indicates the presence of the source organism's DNA, not necessarily the intact, harmful allergen, which can lead to false positives from non-allergenic contaminating material [15] [4].
Table 2: Inherent Advantages and Limitations of ELISA and PCR
| Aspect | ELISA (Protein-Based) | PCR (DNA-Based) |
|---|---|---|
| Analytical Target | Allergenic proteins (the hazard itself) | Species-specific DNA sequences (a marker for the hazard) |
| Quantitative Capability | Strong; directly quantitative [5] | Generally qualitative or semi-quantitative [4] |
| Effect of Food Processing | Proteins may be denatured or altered, leading to potential false negatives [15] [18] | DNA may be fragmented, but short targets are stable; less affected by processing [16] [5] |
| Risk of False Positives | Possible cross-reactivity with similar proteins from non-target sources [4] | Detects DNA from any tissue of the species, which may not correlate with allergenic protein presence [15] |
| Best Suited For | Quantification of allergenic protein, verification of "free-from" claims, regulatory compliance (e.g., gluten) [3] [19] | Detection in highly processed, fermented, or hydrolyzed foods; complex matrices with potential protein interference [3] [5] |
Experimental Workflows and Protocols
ELISA Methodology: Sandwich Assay for Allergen Detection
The sandwich ELISA is the predominant format for allergen detection due to its high specificity and robust quantitative performance [4]. The workflow relies on two antibodies that bind to different epitopes on the same target protein.
Diagram 1: Sandwich ELISA Workflow
Detailed Protocol:
- Protein Extraction: The food sample is homogenized in an appropriate extraction buffer. The buffer composition is critical; high-salt or high-pH buffers are often employed to improve the recovery of proteins that may interact with the food matrix [15] [18]. Optimization of this step is essential for reliable detection, especially in processed foods.
- Capture: The extracted protein solution is added to the wells of a microplate that have been pre-coated with a capture antibody specific to the target allergen (e.g., anti-Ara h 1 for peanut) [5] [4]. Incubation allows the allergen protein to bind to the capture antibody.
- Detection: After washing to remove unbound material, a second enzyme-conjugated antibody (detection antibody), specific to a different epitope on the same allergen, is added. This forms an "antibody-allergen-antibody" sandwich complex [4].
- Signal Development: A colorless enzyme substrate is added. The enzyme (e.g., Horseradish Peroxidase) converts the substrate into a colored product.
- Quantification: The intensity of the color, measured spectrophotometrically, is directly proportional to the concentration of the target allergen in the sample. This is compared against a standard curve prepared with known concentrations of the purified allergen [5].
PCR Methodology: Real-Time PCR for Allergen Source Detection
Real-time PCR (qPCR) detects and amplifies a short, species-specific DNA sequence. The process is monitored in real-time, allowing for the detection of trace amounts of DNA.
Diagram 2: Real-Time PCR Workflow
Detailed Protocol:
- DNA Extraction: DNA is purified from the food sample. The cetyltrimethyl ammonium bromide (CTAB) method is a common and effective approach for plant-based and complex matrices [16]. The goal is to obtain DNA of sufficient purity and length, while inhibiting compounds must be removed.
- Reaction Setup: The extracted DNA is combined with a reaction mix containing primers and a fluorescently labeled probe. The primers are short, single-stranded DNA sequences designed to hybridize specifically to the target gene (e.g., a segment of the Ara h 2 gene for peanut or a glutenin gene for wheat) [16] [17]. The TaqMan probe with a minor groove binder (MGB) provides high specificity [8].
- Amplification & Detection: The mixture undergoes repeated thermal cycles in a real-time PCR instrument:
- Denaturation: High temperature (∼95°C) separates the double-stranded DNA.
- Annealing: Lower temperature (∼60°C) allows primers and probe to bind to the target sequence.
- Extension: The DNA polymerase extends the primers. During this phase, the probe is cleaved, releasing a fluorescent signal. The fluorescence increases with each cycle in proportion to the amount of amplified PCR product [5].
- Analysis: The cycle threshold (Ct), the cycle number at which fluorescence exceeds a background level, is determined. A lower Ct value indicates a higher initial concentration of the target DNA. The result is typically qualitative (presence/absence) or semi-quantitative when compared to reference standards [4].
Research Reagent Solutions and Essential Materials
Successful implementation of ELISA and PCR requires specific, high-quality reagents. The following table catalogues the essential components for each method.
Table 3: Key Research Reagents and Materials for Allergen Detection
| Method | Reagent/Material | Function and Critical Features |
|---|---|---|
| ELISA | Capture & Detection Antibodies | High-affinity, specific antibodies (often monoclonal) that recognize distinct, stable epitopes on the native or denatured allergenic protein. Critical for assay specificity and sensitivity [4]. |
| Protein Extraction Buffers | Optimized buffers (e.g., high-salt, high-pH) to efficiently solubilize allergenic proteins from complex or processed food matrices while preserving epitope integrity [15] [18]. | |
| Enzyme-Substrate System | Conjugated enzyme (e.g., HRP) and its corresponding chromogenic or chemiluminescent substrate. Determines the signal-to-noise ratio and dynamic range of the assay. | |
| Allergen Protein Standards | Purified, quantified allergenic proteins (e.g., Ara h 1, β-lactoglobulin) for generating a standard curve, which is essential for accurate quantification [3]. | |
| PCR | Species-Specific Primers & Probes | Short, synthetic oligonucleotides designed to amplify a unique, short (100-300 bp) sequence from the genome of the allergenic source. Specificity is paramount [8] [16]. |
| DNA Polymerase | Thermostable enzyme (e.g., Taq polymerase) that synthesizes new DNA strands during thermal cycling. Fidelity and processivity affect amplification efficiency. | |
| DNA Extraction Kits/CTAB Reagents | Chemicals and kits for efficient lysis of cells and purification of DNA, free of inhibitors (e.g., polyphenols, polysaccharides) commonly found in food [16]. | |
| dNTPs | Deoxynucleotide triphosphates (dATP, dCTP, dGTP, dTTP), the building blocks for DNA synthesis by the polymerase. | |
| General | Reference Materials | Incurred control samples with known, homogeneously distributed allergen concentrations. Used for method validation to assess recovery and accuracy [4]. |
| Microplates & PCR Plates | Consumables compatible with automated liquid handlers, spectrophotometers, and real-time PCR thermal cyclers. |
The choice between ELISA and PCR is not a question of which method is universally superior, but which is most fit-for-purpose for a specific research or testing scenario. ELISA is the unequivocal choice when the research objective is the direct quantification of specific allergenic proteins, as it measures the causative agent of the allergic reaction itself. It is the established gold standard for compliance with regulatory thresholds, such as for gluten [3] [19]. PCR, however, excels as a highly sensitive detective tool when protein integrity is questionable, such as in highly processed, hydrolyzed, or fermented foods where target epitopes may be destroyed [16] [5]. It is also indispensable for detecting allergens in low-protein matrices like celery or when specific antibodies are unavailable or suffer from cross-reactivity.
For a comprehensive allergen risk assessment, an integrated approach using both methods provides the most robust picture. PCR can serve as a sensitive screening tool to identify the presence of an allergenic source, while ELISA can subsequently quantify the allergenic protein load, offering a complementary strategy that leverages the strengths of both techniques to ensure maximum consumer protection and regulatory compliance [5] [19].
In the field of food allergen detection, the accurate measurement of performance metrics is paramount for method validation and regulatory compliance. Sensitivity, specificity, and limit of detection (LOD) serve as critical benchmarks for evaluating analytical techniques, particularly when comparing established methods like Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR). These parameters determine the reliability of detection systems in identifying trace amounts of allergens in complex food matrices, directly impacting consumer safety and product labeling accuracy. Within the broader thesis comparing ELISA and PCR for allergen detection in processed foods, understanding these metrics provides a foundation for objective methodological evaluation and appropriate technology selection based on specific analytical requirements.
The fundamental distinction between these metrics lies in their respective roles: sensitivity measures a method's ability to correctly identify true positives, specificity indicates its capacity to avoid false positives by correctly identifying true negatives, and LOD represents the lowest analyte concentration that can be reliably detected. For food allergen analysis, where protection of sensitized consumers is the ultimate goal, the precise determination and interpretation of these parameters directly influence methodological choices in both research and quality control environments.
Theoretical Foundations of Performance Metrics
Conceptual Definitions and Distinctions
The terms sensitivity, specificity, and LOD possess precise technical definitions established by international standards organizations, though their application and interpretation vary between immunological and molecular biological methods. The Clinical Laboratory Standards Institute (CLSI) defines LOD as "the lowest amount of analyte in a sample that can be detected with stated probability" [20]. This distinguishes it from the limit of quantification (LOQ), defined as "the lowest amount of measurand that can be quantitatively determined with stated acceptable precision and accuracy" [20]. This distinction is crucial, as detection alone does not ensure precise measurement.
In diagnostic contexts, sensitivity represents the proportion of true positives correctly identified by the assay, while specificity indicates the proportion of true negatives correctly identified [21]. The International Union of Pure and Applied Chemistry (IUPAC) further defines analytical sensitivity as the slope of the calibration curve, representing the method's ability to distinguish small concentration differences [22]. Understanding these nuanced definitions is essential for proper method evaluation and comparison.
Statistical Determination and Calculation Methods
The statistical determination of LOD follows established protocols that differ between linear immunoassays and logarithmic molecular methods. For immunoassays with linear response curves, LOD is typically calculated as LoB + 1.645 × σ low concentration sample, where LoB (limit of blank) equals mean blank + 1.645 × σ blank [20]. This approach assumes normal distribution of data in linear scale and a 95% confidence level.
For qPCR methods with logarithmic response characteristics, standard linear approaches are inappropriate since negative samples produce no Cq values, preventing standard deviation calculation. Instead, logistic regression models based on binary detection outcomes at various concentrations are employed, with maximum likelihood estimation determining the concentration at which 95% of replicates test positive [20]. This method accommodates the fundamental characteristics of molecular amplification data without violating statistical assumptions.
Methodology Comparison: ELISA versus PCR
Fundamental Principles and Mechanisms
ELISA operates on antigen-antibody interaction principles, where antibodies specifically bind to allergenic proteins. In typical sandwich ELISA formats, captured antigens are detected using enzyme-linked secondary antibodies that generate measurable color signals upon substrate addition [5]. The signal intensity correlates with allergen concentration, enabling quantification. The technique's performance depends heavily on antibody affinity, specificity, and the stability of target epitopes through food processing.
PCR-based methods target species-specific DNA sequences rather than proteins. Through cyclic amplification using sequence-specific primers, trace amounts of DNA are exponentially multiplied to detectable levels [5]. Real-time PCR (qPCR) monitors amplification kinetics, providing both qualitative detection and quantitative capability. The method's effectiveness depends on DNA extractability, primer specificity, and the conservation of target sequences across species.
Experimental Protocols for Allergen Detection
Standard ELISA Protocol:
- Sample Preparation: Food samples are homogenized and proteins extracted using appropriate buffers
- Plate Coating: Microplate wells are coated with allergen-specific capture antibodies
- Incubation: Sample extracts are added and allowed to bind during incubation
- Washing: Unbound materials are removed by washing
- Detection: Enzyme-conjugated detection antibodies are added and bind to captured allergens
- Signal Development: Enzyme substrates are added, generating colorimetric signals
- Measurement: Absorbance is measured spectrophotometrically and compared to standards [5]
Standard PCR Protocol:
- DNA Extraction: Food samples are processed to isolate total DNA
- Primer Design: Species-specific primers target unique allergen-encoding sequences
- Amplification: DNA is mixed with primers in a thermal cycler for repeated heating/cooling cycles
- Detection: Amplified DNA is detected in real-time using fluorescent probes or intercalating dyes
- Analysis: Cq values are determined and compared to standard curves for quantification [5]
Recommended Workflows for Complex Matrices
For comprehensive allergen analysis in processed foods, regulatory authorities recommend integrated workflows rather than reliance on single methods. The UK Food Standards Agency advises initial screening with ELISA, followed by confirmatory testing with alternative methods when negative results are obtained [23]. This approach mitigates limitations inherent in individual technologies.
For egg allergen detection, workflows should incorporate multiple ELISA tests targeting different egg white proteins, with LC-MS/MS confirmation [23]. Similarly, milk allergen analysis should target both casein and β-lactoglobulin via ELISA before considering supplemental methods [23]. These workflows acknowledge that no single method currently addresses all analytical challenges across diverse food matrices.
Performance Data Comparison
Quantitative Performance Metrics
Table 1: Direct Comparison of ELISA and PCR Performance Characteristics for Allergen Detection
| Performance Parameter | ELISA | Real-Time PCR |
|---|---|---|
| Dynamic Range | 200-4000 mg/kg [2] | 0.1-106 mg/kg [2] |
| Typical Sensitivity (LOD) | Picograms to nanograms per milliliter [24] | As low as 1-2 viral copies/μL in clinical models [25] |
| Matrix Interference | Significant in complex matrices [2] | Minimal demonstrated [2] |
| Quantitative Capability | Strong within validated range [5] | Limited to qualitative without proper controls [5] |
| Measurement Target | Proteins (allergens directly) [23] | DNA (indirect marker) [23] |
Table 2: Method Application by Allergen Type and Food Matrix
| Allergen/Matrix Scenario | Recommended Method | Rationale | Performance Considerations |
|---|---|---|---|
| Egg, Milk allergens | ELISA [23] [5] | PCR cannot differentiate sources; targets relevant proteins | Prefer kits detecting multiple proteins (casein, β-lactoglobulin for milk) |
| Highly processed foods | PCR [5] | DNA more stable than protein epitopes | May detect non-allergenic material; clinical relevance uncertain |
| Celery, certain fish | PCR [5] | Lack of common antigen for ELISA; low-protein matrix | Currently the only option despite limitations |
| Incident management | ELISA first, then LC-MS/MS confirmation [23] | Avoid false negatives through orthogonal verification | Requires multiple kits targeting different epitopes |
Specificity Considerations in Complex Matrices
Method specificity presents distinct challenges for each technology. ELISA specificity depends on antibody cross-reactivity profiles, which can produce false positives when related non-target proteins share epitopes [23]. PCR specificity is determined by primer design, with potential cross-reactivity to closely related species sharing conserved DNA sequences [5].
In processed foods, matrix effects significantly impact specificity. ELISA demonstrates superior specificity for directly measuring allergenic proteins, the molecules actually responsible for allergic reactions [23]. PCR's indirect measurement through DNA may detect species presence without correlating to allergen content, particularly concerning for ingredients that may contain DNA but not allergenic proteins due to processing or tissue type.
Advanced Detection Technologies
Emerging Methodologies
While ELISA and PCR dominate current allergen detection, several emerging technologies show promise for overcoming their limitations. Mass spectrometry (LC-MS/MS) enables highly specific multiplex detection by targeting proteotypic peptides, providing direct measurement of multiple allergenic proteins simultaneously [23] [26]. Although currently less sensitive than ELISA, its specificity advantages warrant inclusion in confirmatory workflows.
Temperature-responsive liposome-linked immunosorbent assay (TLip-LISA) represents an innovative approach combining liposome technology with immunoassay principles. This method incorporates squaraine dye-containing liposomes that exhibit dramatic fluorescence increase at phase transition temperature, achieving extraordinary sensitivity down to 0.97 aM for PSA detection in model systems [27]. While not yet established for food allergens, this technology demonstrates the potential for significant sensitivity improvements beyond conventional ELISA.
Non-Destructive and Rapid Methods
Hyperspectral imaging (HSI) and Fourier Transform Infrared (FTIR) spectroscopy are emerging as non-destructive alternatives for allergen screening [26]. When combined with machine learning algorithms, these techniques enable real-time monitoring without altering food integrity. Similarly, lateral flow assays (LFAs) provide rapid, on-site testing capabilities suitable for manufacturing environments, though primarily as qualitative screening tools [5].
Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for Allergen Detection Studies
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Reference Materials (RMs) | Method calibration and validation [23] | Preferably incurred materials; limited CRM availability |
| Allergen-Specific Antibodies | Target capture and detection in ELISA [24] | Monoclonal preferred for consistency; affinity critical for sensitivity |
| Species-Specific Primers/Probes | DNA amplification in PCR [5] | Must target unique sequences; design critical for specificity |
| Protein Extraction Buffers | Allergen extraction from food matrices [23] | Composition critical for efficient extraction, especially in processed foods |
| DNA Extraction Kits | Nucleic acid isolation for PCR [5] | Efficiency varies by matrix; must remove PCR inhibitors |
Experimental Workflows
Diagram 1: Comprehensive allergen detection workflow integrating multiple analytical methods for incident management, based on UK Food Standards Agency recommendations [23]. The sequential approach utilizing orthogonal methods reduces false negative risks.
The comparative analysis of ELISA and PCR for food allergen detection reveals a complex landscape where neither technology universally outperforms the other across all applications. Instead, method selection must align with specific analytical requirements, considering the particular allergen, food matrix, processing history, and required sensitivity level. ELISA maintains advantages for directly quantifying allergenic proteins in many applications, while PCR offers superior sensitivity and resistance to matrix effects in challenging scenarios.
The critical evaluation of performance metrics—sensitivity, specificity, and LOD—must extend beyond numerical comparisons to encompass methodological fitness for purpose. Future methodological developments will likely focus on multiplexed detection platforms with enhanced sensitivity and reduced matrix effects, potentially through hybrid approaches combining the complementary strengths of existing technologies. For researchers and food safety professionals, informed method selection based on comprehensive understanding of these performance metrics remains essential for effective allergen management and consumer protection.
Food allergies represent a significant and growing public health concern worldwide, with strict avoidance of allergenic foods being the primary management strategy for affected individuals. This reality places immense importance on accurate allergen detection and clear food labeling to ensure consumer safety. The global allergen testing market is experiencing substantial growth, driven by increasing allergy prevalence, stringent regulatory requirements, and technological advancements in detection methodologies. Within this landscape, ELISA (Enzyme-Linked Immunosorbent Assay) and PCR (Polymerase Chain Reaction) have emerged as two dominant analytical techniques, each with distinct advantages and limitations for detecting allergens in processed foods. This guide provides an objective comparison of these methodologies, focusing on their performance characteristics, applications in complex food matrices, and positioning within the current regulatory framework.
The global food allergen testing market is positioned for significant expansion, with its value projected to increase from USD 970.3 million in 2025 to USD 2,062.6 million by 2035, reflecting a compound annual growth rate (CAGR) of 7.8% [28]. This growth trajectory is mirrored in the broader allergy diagnostics sector, which includes clinical applications and is expected to grow from US$5.8 billion in 2024 to US$10.7 billion by 2030, at a CAGR of 10.8% [29].
Several key factors are propelling this market expansion:
- Rising Allergy Prevalence: Increasing incidence of food allergies worldwide, particularly among children [28]
- Stringent Regulations: Implementation of stricter food safety laws and labeling requirements across regions [19] [28]
- Consumer Awareness: Growing consumer demand for allergen-free products and transparent labeling [29]
- Technological Innovation: Development of more sensitive, rapid, and multiplexed detection platforms [26] [28]
Table 1: Global Food Allergen Testing Market Outlook
| Metric | 2025 Value | 2035 Projected Value | CAGR (2025-2035) |
|---|---|---|---|
| Market Size | USD 970.3 million | USD 2,062.6 million | 7.8% |
| Technology Leader | PCR-based Testing (35.4% share) | ||
| Top Application | Processed Food (28% share) | ||
| Leading Source | Milk (25% share) |
Regionally, North America currently dominates the allergy diagnostics market, holding an estimated 45.4% share in 2024 [29]. However, the Asia-Pacific region is projected to record the fastest growth rate in allergy diagnostics, with a CAGR of 11.8% during 2024-2030, driven by rapid urbanization, increasing pollution, and healthcare infrastructure development [29]. Japan leads growth projections for food allergen testing with a CAGR of 6.5% from 2025 to 2035, followed by the UK (6.2% CAGR) and Germany (6.1% CAGR) [28].
Regulatory Framework
The regulatory landscape for allergen management has evolved significantly to protect consumer health. In the United States, the Food Allergen Labeling and Consumer Protection Act (FALCPA) identifies nine major food allergens: milk, eggs, fish, Crustacean shellfish, tree nuts, peanuts, wheat, soybeans, and sesame [30]. The Food and Drug Administration (FDA) enforces labeling requirements for these allergens in most packaged foods, requiring clear declaration of allergen sources either in the ingredient list or through a "contains" statement [30].
A critical regulatory aspect is that the FDA has not established threshold levels for any allergens, meaning there is no defined value below which allergen presence is considered safe for all allergic individuals [30]. This regulatory stance places additional responsibility on food manufacturers to implement stringent controls and sensitive detection methods to prevent cross-contamination.
Globally, regulatory approaches vary, with regions like the European Union implementing the Food Information for Consumers Regulation, and countries like Japan maintaining strict allergen declaration frameworks with defined thresholds (10 μg/g for certain allergens) [3]. These regulatory differences create compliance complexities for global food manufacturers, necessitating versatile testing approaches that can meet varying regional requirements.
Methodology Comparison: ELISA vs. PCR
Principles and Technologies
ELISA (Enzyme-Linked Immunosorbent Assay) is an immunochemical method that detects allergenic proteins through antigen-antibody interactions. The process involves:
- Sample Preparation: Proteins are extracted from food samples using buffer solutions, often at high temperatures to release potential allergens [13]
- Antibody Coating: A microplate is pre-coated with antibodies specific to the target allergen [5] [13]
- Binding Phase: Allergenic proteins in the sample bind to the capture antibodies [5]
- Detection: A second enzyme-linked antibody binds to the captured allergen [5] [13]
- Signal Measurement: Enzyme substrate addition produces a color change measurable via spectrophotometry, with intensity proportional to allergen concentration [5] [13]
PCR (Polymerase Chain Reaction) is a molecular biology technique that amplifies specific DNA sequences associated with allergenic foods. The methodology includes:
- DNA Extraction: Isolation of DNA from food samples using various extraction protocols [16]
- Amplification: Target DNA sequences are amplified using specific primers in a thermal cycler [5]
- Detection: Amplified DNA is detected via real-time PCR, providing measurable signals indicating allergen presence [5] [16]
Experimental Performance Data
Recent studies provide quantitative performance data for both techniques in processed food matrices:
Table 2: Methodological Comparison of ELISA and PCR for Allergen Detection
| Parameter | ELISA | PCR |
|---|---|---|
| Target Molecule | Proteins (direct detection) | DNA (indirect detection) |
| Sensitivity | Parts per million (ppm) levels; High sensitivity for native proteins [19] | High sensitivity for target DNA sequences; Effective even in processed foods [16] |
| Specificity | High; Dependent on antibody quality [19] | High; Primers target unique allergen gene sequences [16] |
| Quantification | Direct quantitative results [5] | Primarily qualitative; Semi-quantitative with standard curves [5] |
| Impact of Processing | Protein denaturation may reduce detectability [3] [19] | DNA stability allows detection in processed foods [3] [16] |
| Detection Time | ~30 minutes to several hours [13] | Several hours including DNA extraction [5] |
| Multiplexing Capability | Limited | High with multiplex PCR [28] |
| Regulatory Status | Gold standard for routine screening [19] | Official method in some countries (e.g., Germany, Japan) [3] |
Research on wheat and maize allergen detection demonstrates PCR's effectiveness in processed foods, where DNA remains amplifiable even after baking at 220°C for 40-60 minutes, though with some degradation [16]. For reliable PCR analysis of processed foods, target amplicons should be limited to approximately 200-300 base pairs to accommodate potential DNA fragmentation [16].
ELISA maintains advantages for certain applications, being the officially recognized method for gluten detection by the Codex Alimentarius, with a defined threshold of 20 mg/kg [3]. The technique's direct measurement of allergenic proteins rather than genetic markers provides more clinically relevant data for allergy risk assessment.
Allergen Detection Method Selection Workflow
Application in Processed Foods
The selection between ELISA and PCR becomes particularly significant when analyzing processed foods, where manufacturing operations can alter the detectability of target molecules. Thermal processing, fermentation, hydrolysis, and high-pressure treatments can denature proteins, potentially affecting antibody recognition in ELISA methods [3] [19]. Conversely, DNA demonstrates greater stability through various food processing conditions, making PCR particularly valuable for detecting allergens in baked goods, extruded snacks, hydrolyzed products, and fermented foods [5] [19].
Research demonstrates that while genomic DNA undergoes degradation during high-temperature processing (e.g., baking at 180-220°C), appropriate primer design targeting shorter DNA fragments (200-300 bp) maintains reliable detection of wheat and maize allergens [16]. This robustness makes PCR particularly suitable for verifying allergen-free claims in complex processed products where protein integrity may be compromised [5].
ELISA maintains advantages for detecting specific allergenic proteins in native or minimally processed matrices, and remains the preferred method when quantitative protein data is required for compliance with specific regulatory standards, such as gluten-free labeling [5] [3]. The direct measurement of allergenic proteins rather than genetic markers provides more clinically relevant information about potential allergenicity.
Emerging Technologies and Future Outlook
The allergen testing landscape is evolving with several promising technologies emerging to address current methodological limitations:
- Mass Spectrometry: Particularly liquid chromatography-mass spectrometry (LC-MS), enables highly specific detection and quantification of multiple allergenic proteins simultaneously through identification of proteotypic peptides, offering high precision across complex food matrices [26]
- Biosensors: Various biosensor platforms are under development, leveraging aptamers, antibodies, or cell-based recognition elements coupled with electrochemical, optical, or piezoelectric transducers for rapid, sensitive allergen detection [3]
- AI-Enhanced Platforms: Integration of artificial intelligence with detection technologies like hyperspectral imaging, Fourier Transform Infrared (FTIR) spectroscopy, and computer vision enables non-destructive, real-time allergen monitoring without compromising food integrity [26]
- Digital ELISA and Microfluidics: Advanced immunoassay formats offering improved sensitivity through single-molecule counting and reduced sample/reagent requirements, with recent developments demonstrating 60% reduced sample volumes (20 μL vs. 50 μL) compared to conventional systems [31]
These emerging technologies promise to address current challenges related to detection sensitivity, multiplexing capability, analysis time, and applicability to complex matrices. The integration of cloud-based data management platforms further enhances utility by providing centralized dashboards for compliance documentation and trend analysis [26].
Essential Research Reagent Solutions
Successful implementation of allergen testing methodologies requires specific reagent systems and materials:
Table 3: Essential Research Reagents for Allergen Detection
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Allergen-Specific Antibodies | Recognition and binding to target allergenic proteins | Critical for ELISA specificity; monoclonal antibodies reduce cross-reactivity [13] |
| Protein Extraction Buffers | Solubilize and extract proteins from food matrices | Composition optimized for different food types; may include denaturants or reducing agents [13] |
| DNA Extraction Kits | Isolate DNA from complex food matrices | CTAB-based methods effective for plant-derived foods [16] |
| Species-Specific Primers | Amplify target DNA sequences in PCR | Designed for conserved allergen genes; short amplicons (200-300 bp) for processed foods [16] |
| Microplates and Readout Systems | Solid phase for binding and detection | Spectrophotometers for colorimetric ELISA; thermal cyclers and fluorescence detectors for PCR [5] |
| Reference Materials | Method validation and calibration | Certified reference materials with known allergen content essential for quantification [3] |
The current market and regulatory landscape for allergen testing reflects a dynamic field balancing technological innovation with increasing public health demands. While ELISA remains the gold standard for routine allergen screening due to its direct protein detection, cost-effectiveness, and regulatory acceptance, PCR technology has established a crucial complementary role, particularly for processed foods where protein denaturation may compromise immunoassay effectiveness. The choice between these methodologies depends on multiple factors, including food matrix composition, degree of processing, regulatory requirements, and needed output (quantitative vs. qualitative). Future methodological developments will likely focus on multiplexing capabilities, rapid on-site testing, enhanced sensitivity, and integration with data analytics platforms to address the evolving needs of food manufacturers, regulatory agencies, and allergic consumers.
Strategic Application: Selecting ELISA or PCR Based on Food Matrix and Processing
ELISA as the Gold Standard for Quantitative Protein Analysis in Raw Ingredients
Among the various analytical techniques available for allergen detection, the Enzyme-Linked Immunosorbent Assay (ELISA) maintains its position as the gold standard for quantitative protein analysis in raw food ingredients. This comprehensive guide examines the technical foundations, performance parameters, and practical applications of ELISA methodology in comparison with emerging molecular techniques, particularly Polymerase Chain Reaction (PCR). Through systematic evaluation of experimental data and validation criteria, we demonstrate why ELISA remains the preferred platform for precise protein quantification in research, quality control, and regulatory compliance contexts within food safety and pharmaceutical development.
Food allergen detection has become increasingly critical in public health and food safety, with undeclared allergens consistently representing the leading cause of food product recalls globally [4]. Accurate detection and quantification of allergenic proteins in raw ingredients establishes the foundation for effective allergen control programs, compliance with labeling regulations, and protection of consumer health. The two predominant analytical approaches for allergen detection target different molecular entities: immunoassays detect allergenic proteins directly, while molecular methods detect species-specific DNA sequences [5] [3].
Within this landscape, ELISA has emerged as the benchmark technique for quantitative protein analysis due to its robust antigen-antibody interaction principle, standardized protocols, and well-characterized validation parameters [32]. The technique's direct measurement of clinically relevant proteins—the molecules that actually trigger allergic responses—provides a significant advantage over indirect DNA-based methods when determining the potential allergenicity of food ingredients [4]. Internationally recognized bodies including the Codex Alimentarius Commission have formally adopted ELISA as the official reference method for detecting specific allergens like gluten in foods [3].
Technical Comparison of ELISA and PCR Methodologies
Fundamental Principles and Workflows
ELISA operates on the principle of specific antigen-antibody recognition, typically employing a sandwich format where the target protein is captured between two antibodies [4]. This double-antibody recognition system provides exceptional specificity, while enzyme-mediated signal amplification enables sensitive quantification of the target protein [5]. The direct measurement of proteins makes ELISA particularly valuable for assessing the actual allergenic risk in food products.
PCR technology amplifies specific DNA sequences unique to allergenic species using thermal cycling and DNA polymerase enzymes [5]. While exceptionally sensitive for detecting species-specific genetic material, PCR does not directly measure the allergenic proteins themselves, potentially leading to discrepancies between detected DNA and actual allergenicity [33].
The experimental workflow for each method differs significantly, as illustrated below:
Comparative Performance Characteristics
The table below summarizes the fundamental operational differences between ELISA and PCR methods:
| Parameter | ELISA | PCR |
|---|---|---|
| Detection Target | Specific allergenic proteins [4] | Species-specific DNA sequences [5] |
| Measurement Output | Quantitative protein concentration [5] [34] | Qualitative or semi-quantitative DNA detection [5] [4] |
| Sensitivity | High (parts per million range) [35] | Very high (detects trace DNA) [4] |
| Specificity | Protein epitope specificity [36] | Species-level DNA specificity [4] |
| Impact of Food Processing | Protein structure may be denatured [5] | DNA is more stable through processing [5] |
| Regulatory Status | Official Codex method for gluten [3] | Official method in Germany and Japan [3] |
Experimental Data and Validation Metrics
ELISA Performance Validation Parameters
Rigorous validation establishes the reliability of quantitative ELISA methods. The following validation parameters demonstrate why ELISA meets gold standard status for protein quantification:
Precision and Reproducibility: Intra-assay precision (within-plate variability) typically shows coefficient of variation (CV) <10%, while inter-assay precision (between-plate variability) also maintains CV <10% across different operators and days [36]. This consistency ensures reproducible results in quality control environments.
Accuracy and Recovery: Spike-and-recovery experiments evaluate accuracy by measuring the detection of known analyte quantities added to sample matrices. Recovery rates of 80-120% indicate minimal matrix interference [36]. For sesame protein detection in incurred foods, recovery rates of 67-81% demonstrate acceptable performance in complex matrices [35].
Sensitivity and Specificity: ELISA methods exhibit exceptional sensitivity with limits of detection (LOD) as low as 0.013 μg/g for sesame proteins [35]. Specificity is ensured through antibodies that recognize target proteins without cross-reactivity to related molecules, validated through comprehensive cross-reactivity panels [36] [32].
The quantitative performance of ELISA is demonstrated in the following validation data:
| Validation Parameter | Typical Performance | Experimental Example |
|---|---|---|
| Intra-Assay Precision | CV <10% [36] | VCAM-1 Human ELISA: CV 4.85-7.68% across samples [36] |
| Inter-Assay Precision | CV <10% [36] | Amyloid beta 42 ELISA: CV 5.32-9.85% across multiple runs [36] |
| Linearity of Dilution | 70-130% of expected [36] | c-Myc Human ELISA: 76-126% across serial dilutions [36] |
| Recovery Rate | 80-120% [36] | Sesame protein ELISA: 67-81% in incurred foods [35] |
| Limit of Detection | Varies by target | Sesame protein ELISA: 0.013 μg/g [35] |
Comparative Method Performance Data
Direct comparison studies reveal contextual advantages for each method. In malaria research, a mitochondrial COX-I PCR method demonstrated higher sensitivity (67-88% detection in abdomen segments) compared to CSP ELISA (single detection) during early infection stages in mosquitoes [33]. However, this superior sensitivity comes with a significant limitation—the PCR method detected all parasite life stages rather than specifically identifying infectious sporozoites [33].
For food allergen applications, ELISA provides direct measurement of clinically relevant proteins at regulated thresholds. The Codex Alimentarius specifies ELISA as the method for gluten detection with a regulatory threshold of 20 mg/kg, while Japan implements both ELISA and PCR with an allergen threshold of 10 μg/g [3].
Method Selection Framework
The decision between ELISA and PCR methods depends on multiple experimental factors, which can be navigated using the following workflow:
Application-Specific Recommendations
ELISA is Recommended For:
- Quantitative analysis of allergenic proteins in raw ingredients [5]
- Regulatory compliance and verification of "free-from" claims [4]
- Process validation where protein detection correlates directly with allergenicity [3]
- Applications requiring precise quantification against established thresholds [35]
PCR is Preferred For:
- Detection of allergenic species in highly processed foods where proteins may be denatured [5]
- Identification of specific biological species in complex matrices [3]
- Situations where DNA stability offers advantage over protein detectability [5]
- Qualitative screening when extreme sensitivity is required [33]
Combined Approaches are particularly powerful in complex scenarios, such as:
- Method verification and confirmatory analysis [5]
- Comprehensive allergen control programs [4]
- Research applications characterizing both presence and potency of allergens [3]
Research Reagent Solutions and Experimental Implementation
Essential Components for ELISA
Successful implementation of quantitative ELISA requires specific reagent systems and laboratory materials:
| Reagent/Material | Function | Application Notes |
|---|---|---|
| Capture Antibodies | Bind target protein to solid phase [32] | High affinity, specific to target epitope; require concentration optimization [32] |
| Detection Antibodies | Recognize different epitope on captured protein [4] | Typically enzyme-conjugated (HRP, AP); enable signal generation [36] |
| Blocking Buffers | Prevent non-specific binding [32] | BSA, non-fat dry milk, or commercial formulations; require optimization [32] |
| Enzyme Substrates | Generate detectable signal [36] | Colorimetric, chemiluminescent, or fluorescent; selection depends on sensitivity needs [36] |
| Reference Standards | Calibrate quantitative measurements [34] | Certified reference materials calibrated to international standards (e.g., NIBSC) [36] |
| Microplate Readers | Measure signal intensity [36] | Spectrophotometers for colorimetric detection; compatible with 96-well format [36] |
Standard ELISA Protocol for Protein Quantification
The following detailed methodology ensures reliable quantification of allergenic proteins in raw ingredients:
Sample Preparation:
- Homogenize representative samples of raw ingredients using appropriate extraction buffers [35]
- Extract proteins using buffers optimized for target allergens and matrix compatibility [32]
- Clarify extracts by centrifugation (10,000 × g for 10 minutes at 4°C)
- Determine optimal sample dilution to fall within assay dynamic range [34]
Assay Procedure:
- Coat microplate wells with capture antibody (typically 1-10 μg/mL in coating buffer) and incubate overnight at 4°C [32]
- Block remaining protein-binding sites with appropriate blocking buffer (1-2 hours at room temperature) [32]
- Add standards and samples to wells in duplicate or triplicate; incubate 1-2 hours at room temperature [34]
- Wash plates thoroughly (3-5 times with wash buffer) to remove unbound proteins [32]
- Add enzyme-conjugated detection antibody; incubate 1-2 hours at room temperature [36]
- Repeat washing step to remove unbound detection antibody [32]
- Add enzyme substrate solution; incubate for precise duration (typically 15-30 minutes) [36]
- Stop reaction (if required) and measure signal intensity using microplate reader [34]
Data Analysis:
- Generate standard curve using reference standards of known concentration [34]
- Apply 4-parameter logistic (4PL) curve fit for optimal quantitative accuracy [34]
- Interpolate sample concentrations from standard curve [34]
- Apply dilution factors to calculate original sample concentration [34]
- Validate assay acceptance criteria (R² > 0.99 for standard curve, CV < 10% for replicates) [36]
ELISA maintains its gold standard status for quantitative protein analysis in raw ingredients through its direct measurement of clinically relevant allergens, robust validation parameters, and regulatory acceptance. While PCR offers advantages in specific scenarios—particularly for detecting allergenic species in highly processed foods—ELISA remains unmatched for precise protein quantification essential for regulatory compliance and consumer protection. The methodological framework presented here enables researchers to implement ELISA methodologies effectively while understanding the complementary role of PCR-based approaches in comprehensive allergen detection strategies. As food safety regulations evolve globally, the quantitative precision of ELISA ensures its continued relevance in protecting allergic consumers through accurate allergen detection and quantification.
For researchers and drug development professionals, accurately detecting food allergens is a critical component of food safety and regulatory compliance. The challenge intensifies with processed foods, where heating, fermentation, and other technological treatments can alter the native structure of allergenic proteins, compromising detection accuracy. Within this context, two methodological approaches dominate the field: the protein-based Enzyme-Linked Immunosorbent Assay (ELISA) and the DNA-based Polymerase Chain Reaction (PCR). This guide provides an objective, data-driven comparison of these technologies, focusing on their performance in analyzing highly processed foods where the stability of DNA versus protein becomes a decisive factor. The core thesis is that PCR offers distinct advantages for processed food analysis due to the superior stability of the DNA molecule under conditions of heat and fermentation that often denature protein epitopes targeted by ELISA.
Technical Comparison: PCR vs. ELISA Fundamentals
ELISA and PCR employ fundamentally different detection principles, which underpins their varying performance in different food matrices.
ELISA (Enzyme-Linked Immunosorbent Assay): This immunoassay detects specific allergenic proteins (the antigens) using antibody-antigen interactions. In the most common format for allergen detection, the sandwich ELISA, a capture antibody immobilized on a plate binds the target protein from the sample extract. A second, enzyme-linked detection antibody then binds to a different epitope on the same protein, forming a "sandwich." The subsequent addition of an enzyme substrate produces a colorimetric signal proportional to the amount of protein present [5] [4]. The method's effectiveness is therefore directly dependent on the structural integrity of the protein and its antibody-binding epitopes.
PCR (Polymerase Chain Reaction): This molecular technique detects the DNA of the species containing the allergen. It uses primers to target and amplify a unique, species-specific DNA sequence via thermal cycling. The amplification process is monitored in real-time (qPCR), with the signal intensity correlating with the amount of target DNA initially present in the sample [5] [4]. PCR does not detect the allergen itself but rather the genetic material of the allergenic source, which is often more resilient to processing.
The following workflow illustrates the key steps of the real-time PCR process for allergen detection:
Experimental Data: Direct Performance Comparison
A direct, side-by-side comparative study of PCR and ELISA for detecting crustacean shellfish allergens provides compelling quantitative evidence for PCR's advantages. The study used identical split samples across two different food matrices—Manhattan clam chowder and fish sauce—and evaluated four real-time PCR methods alongside two commercial ELISA kits [2].
Key Findings from the Comparative Study:
- Dynamic Range: The PCR assays demonstrated a significantly broader dynamic range (0.1–100,000 mg/kg) compared to the ELISA assays, which had a much narrower range (200–4,000 mg/kg) [2].
- Matrix Interference: The PCR methods did not show significant matrix interference, whereas the ELISA assays were susceptible to interference from the complex food matrices [2].
- Qualitative Agreement: In cases where ELISA assays functioned without matrix interference, there was good qualitative agreement between the two methodologies, confirming that both are reliable for detecting unprocessed allergens [2].
Further evidence comes from the development of a novel ELISA for soy allergens in processed foods. This research highlighted that heat processing significantly influences protein recovery by ELISA. The recovery rate decreased as the intensity of heat treatment increased, a problem that was far less pronounced for PCR-based methods [37].
Table 1: Comparative Performance of PCR and ELISA in Processed Foods
| Performance Characteristic | PCR | ELISA |
|---|---|---|
| Target Molecule | DNA | Protein |
| Dynamic Range | 0.1 - 106 mg/kg [2] | 200 - 4,000 mg/kg [2] |
| Effect of Heat Processing | Minimal (DNA is stable) | Significant (Proteins denature) [37] |
| Matrix Interference | Low [2] | Observed in complex matrices [2] |
| Detection in Sterilized Pâté | Effective | Ineffective (Competitive ELISA failed) [37] |
| Quantification | Semi-quantitative [4] | Fully quantitative [5] [4] |
The Core Advantage: DNA Stability in Processing
The data clearly point to one underlying cause for PCR's superiority in processed foods: the inherent stability of DNA compared to protein.
Food processing techniques—including thermal treatment (pasteurization, sterilization), high-pressure processing, and fermentation—cause structural denaturation, chemical modification, and aggregation of allergenic proteins [38]. These alterations can destroy or mask the epitopes recognized by ELISA's antibodies, leading to false negatives or significant under-reporting of allergen content [37] [4]. For instance, a study on soy detection found that a sandwich ELISA could detect 10 μg/g of soy isolate in pasteurized sausage, but this sensitivity dropped to 1000 μg/g in sterilized pâté, while a competitive ELISA failed to detect any glycinin in the same pâté samples [37].
In contrast, DNA is a more stable molecule that remains detectable even after severe processes that denature proteins. While DNA can be fragmented by extreme conditions, short, targetable sequences often survive. PCR primers are designed to amplify these short, specific fragments, making the method exceptionally robust for detecting allergens in baked goods, hydrolyzed and fermented products, and sterilized foods where ELISA struggles [5] [4]. This stability is also why PCR is the preferred method for detecting allergens in fermented foods, where active microbial processes can further degrade protein structures [5].
The following chart conceptualizes how the detectability of proteins and DNA changes with increasing processing intensity:
Experimental Protocols for Method Validation
For researchers seeking to implement or validate these methods, understanding the core experimental protocols is essential.
Real-Time PCR Protocol for Allergen Detection
A validated real-time PCR protocol for specific detection, as described in a study on probiotic stability, can be adapted for allergen work [39]. The reaction mixture and cycling conditions are detailed below.
Table 2: Example Real-Time PCR Reaction Setup
| Component | Volume per Reaction | Function |
|---|---|---|
| 2x SensiFast Probes Master Mix | 10.0 μL | Provides nucleotides, buffer, polymerase, and probe-optimized components. |
| Forward Primer (10 μM) | 1.8 μL | Binds to one strand of the target DNA sequence. |
| Reverse Primer (10 μM) | 1.8 μL | Binds to the complementary strand of the target DNA. |
| Hydrolysis Probe (5 μM) | 1.0 μL | Binds specifically to the target amplicon, providing fluorescence. |
| DNA Template | 1.0 μL | The extracted sample DNA containing the target sequence. |
| Molecular Biology Grade Water | Up to 20 μL | Brings the reaction to the final volume. |
Thermal Cycling Conditions:
- Initial Denaturation: 95°C for 5 minutes. (Activates the polymerase and denatures the DNA.)
- Amplification (40 cycles):
- Denaturation: 95°C for 10 seconds.
- Annealing/Extension: 60°C for 20 seconds. (Primers bind and the polymerase extends the DNA strand.)
- Data Collection: Fluorescence is measured at the end of each annealing/extension step [39].
Sample Preparation and Validation Considerations
- DNA Extraction: The initial and most critical step for PCR. Commercial kits, such as the NucleoSpin Food kit, are commonly used to efficiently isolate DNA from complex food matrices, removing inhibitors that could affect the PCR reaction [39].
- Method Specificity and Sensitivity: Before application, any PCR assay must be validated for specificity (ensuring no cross-reactivity with non-target species) and sensitivity (determining the Limit of Detection (LOD)). This involves testing against a panel of target and non-target DNA [39].
- Matrix Effects: The performance of the method (both PCR and ELISA) should be validated in the specific food matrix being tested, as components like fats, polyphenols, and salts can interfere with analyses [4].
Research Reagent Solutions
For laboratories establishing or running allergen detection protocols, the following reagents and tools are essential.
Table 3: Essential Research Reagents and Tools for Allergen Detection
| Item | Function in Research |
|---|---|
| Species-Specific Primers & Probes | Short, custom-designed DNA sequences that define the specificity of the PCR assay by binding to and detecting unique genetic markers of the allergenic source. |
| DNA Extraction Kit (e.g., NucleoSpin Food Kit) | Standardized system for efficient purification of inhibitor-free DNA from complex food matrices, critical for reproducible PCR results. |
| Real-Time PCR Master Mix | Optimized buffer containing thermostable DNA polymerase, dNTPs, and salts, often including reference dyes for simplified reaction setup. |
| Protein Extraction Solution | Validated solutions for efficiently solubilizing allergenic proteins from food samples, crucial for both ELISA and LC-MS/MS analysis. |
| Allergen-Specific Antibodies (Monoclonal/Polyclonal) | The core of ELISA tests, these provide the high specificity needed to bind to and detect target allergenic proteins amidst other food components. |
| HRAM-LC-MS/MS System | High-resolution mass spectrometry system used for confirmatory multi-allergen analysis, especially useful when protein modifications are suspected. |
The experimental data and methodological comparisons robustly support the thesis that PCR holds a superior position for detecting allergens in highly processed foods. The core advantage lies in the fundamental stability of the DNA molecule, which withstands the heat, pressure, and fermentation processes that denature proteins and compromise ELISA's accuracy. PCR's broader dynamic range and resistance to matrix effects further solidify its utility for complex, processed products.
However, a holistic food allergen management strategy recognizes that no single method is universally superior. While PCR excels in the scenarios discussed, ELISA remains the gold standard for quantitative analysis of native proteins in less processed matrices and is the prescribed method for regulatory purposes like gluten analysis [5] [4]. The emerging trend is toward an integrated approach. For instance, combining the rapid screening capability of PCR with the protein-level confirmation of ELISA or the high specificity of Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) provides the most comprehensive safety assurance [5] [40]. For researchers and drug developers, the choice between PCR and ELISA must be guided by the specific food matrix, the nature of the processing involved, and the required analytical outcome.
For researchers and drug development professionals working in food safety, selecting the appropriate analytical method for allergen detection is paramount. The choice between Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR) is not one-size-fits-all; it is profoundly influenced by the food matrix under investigation. Proteins, the molecules that actually trigger allergic reactions, are the direct target of ELISA methods. In contrast, PCR techniques target the DNA of the allergenic species, providing an indirect measure of potential allergen presence [5] [4]. The central challenge for scientists lies in the fact that various matrices—from dairy products to thermally processed baked goods—differentially affect the stability and detectability of these target molecules. This guide provides a detailed, evidence-based comparison of ELISA and PCR, offering matrix-specific protocols and performance data to inform robust experimental design in food allergen research.
Fundamental Principles and a Direct Comparison
Understanding the core mechanisms of ELISA and PCR is essential for interpreting their performance across different environments. ELISA operates on an immunological principle. It uses antibodies to specifically bind and detect allergenic proteins (the antigens) [5]. The most common format for allergen detection is the sandwich ELISA, which employs two antibodies for high specificity, resulting in a colorimetric signal that is quantitative and correlates with the protein concentration [4]. PCR, a molecular technique, detects the DNA of the allergenic source. It amplifies specific, unique DNA sequences to detectable levels, making it exceptionally sensitive to trace amounts of genetic material, though it is generally considered qualitative or semi-quantitative [5] [4].
The table below summarizes the core characteristics of these two techniques.
Table 1: Fundamental comparison of ELISA and PCR methodologies for allergen detection.
| Characteristic | ELISA (Enzyme-Linked Immunosorbent Assay) | PCR (Polymerase Chain Reaction) |
|---|---|---|
| Target Molecule | Allergenic proteins (the actual elicitor) [4] | Species-specific DNA (an indirect marker) [4] |
| Detection Principle | Antigen-antibody binding with enzymatic color development [5] | Amplification of target DNA sequences [5] |
| Nature of Result | Quantitative (e.g., ppm of protein) [5] [4] | Primarily qualitative or semi-quantitative [4] |
| Key Strength | Directly measures the allergenic component; well-standardized for quantitation [19] | High sensitivity; DNA stability allows detection in processed foods where proteins are degraded [5] [19] |
| Key Limitation | Protein denaturation in processed foods can lead to false negatives [41] [18] | Does not detect the allergenic protein itself; result can be influenced by DNA extraction efficiency [4] |
Matrix-Specific Performance and Experimental Data
The food matrix presents unique challenges that can significantly impact method performance. The following section provides experimental data and guidance for key matrix categories.
Dairy Matrices
In dairy products like chocolate, ice cream, or yogurt, fats and proteins can interfere with analyte detection. ELISA is generally the preferred method for quantifying milk allergens (e.g., casein, β-lactoglobulin) because it directly measures the offending proteins [5] [42]. However, PCR can serve as a complementary tool for verifying the presence of species-specific ingredients in complex dairy blends.
- Supported Experimental Data: A study evaluating LC-MS/MS noted that ELISA could reliably detect milk allergens, but also highlighted that in some processed materials, even levels as high as 1000 mg/kg could be missed, underscoring the need for method validation in the specific matrix [41]. Furthermore, newer ELISA kits have been developed with optimized extraction buffers to improve protein recovery from complex matrices like chocolate and ice cream, achieving limits of quantification (LoQ) for β-lactoglobulin as low as 0.031 mg/kg [18].
Baked Goods and Heavily Processed Foods
Thermal processing is a critical factor. Heat can denature proteins, altering the epitopes that ELISA antibodies recognize, which leads to underestimated allergen content or false-negative results [19] [18]. Conversely, DNA is more stable under heat and pressure, making PCR a powerful tool for this matrix.
- Supported Experimental Data: Research comparing real-time PCR and ELISA for crustacean allergens in complex matrices found that PCR had a broader dynamic range (0.1–106 mg/kg) and showed less matrix interference compared to ELISA [2]. In a model study using incurred bread, ELISA failed to detect egg, soy, and milk allergens at concentrations of 1000 mg/kg after the baking process, whereas a mass spectrometric method successfully identified them [41]. This evidence strongly supports the use of PCR as a confirmatory method in thermally processed products.
Complex Matrices (Spices, Chocolate, Meat)
Spices, chocolate, and cooked meats contain compounds that can inhibit enzymatic reactions or bind to target molecules. PCR's high sensitivity makes it particularly suited for detecting low levels of allergens in these challenging environments [42].
- Supported Experimental Data: Proficiency testing has demonstrated that PCR methods provide reliable qualitative detection of allergens like soy and gluten across various matrices, including pastry and sausage meat [43]. For allergens like celery and mustard, which are low-protein matrices or have issues with antibody cross-reactivity, PCR is often the more reliable choice [5]. Commercial PCR kits have been validated to detect allergens at levels as low as 0.1–1 ppm in complex matrices such as spices, chocolate, and cooked meats [42].
Table 2: Summary of recommended methods and performance data for different food matrices.
| Food Matrix | Recommended Method | Key Experimental Findings | Considerations for Researchers |
|---|---|---|---|
| Dairy (e.g., chocolate, ice cream) | ELISA (for milk protein quantitation) | New ELISA kits achieve a LoQ of 0.031 mg/kg for β-lactoglobulin [18]. | Use validated ELISA kits with optimized protein extraction protocols. |
| Baked Goods | PCR (as a confirmatory method) | PCR detected allergens in baked bread where ELISA failed at 1000 mg/kg [41]. | PCR is superior for detecting thermally processed ingredients; ELISA may underestimate risk. |
| Complex Matrices (e.g., spices, meats) | PCR (for high sensitivity) | PCR kits reliably detect allergens down to 0.1–1 ppm in spices and cooked meats [42]. | Effective DNA extraction is critical to overcome PCR inhibitors naturally present in these matrices. |
Experimental Protocols for Key Workflows
To ensure reproducible and reliable results, researchers must adhere to robust and standardized protocols. The diagrams below outline the core workflows for ELISA and PCR.
ELISA Workflow for Protein Allergen Detection
The following chart visualizes the key steps in a sandwich ELISA procedure, which is the dominant format for allergen testing due to its high specificity [4].
Figure 1: Key steps in a sandwich ELISA procedure.
Detailed Protocol:
- Protein Extraction: Homogenize the food sample using a suitable extraction buffer. The buffer is designed to solubilize proteins while minimizing interference from fats, carbohydrates, and other matrix components. Centrifuge to clarify the extract [5] [18].
- Add to Antibody-Coated Well: Transfer the protein extract to a microplate well that has been pre-coated with a capture antibody specific to the target allergen (e.g., anti-Ara h 1 for peanut). Incubate to allow the allergen protein to bind [5] [4].
- Add Detection Antibody: After washing, add an enzyme-linked detection antibody that binds to a different epitope on the captured allergen protein, forming an "antibody-allergen-antibody" sandwich [4].
- Add Enzyme Substrate: Wash again to remove unbound detection antibody. Add a substrate solution that the enzyme converts into a colored product [5].
- Measure Signal: Measure the color intensity (optical density) using a spectrophotometric microplate reader. The intensity is directly proportional to the concentration of the allergen in the sample, which is determined by comparison to a standard curve [5] [4].
PCR Workflow for DNA Allergen Detection
This chart illustrates the fundamental steps in a real-time PCR (qPCR) assay for allergen detection.
Figure 2: Key steps in a PCR detection procedure.
Detailed Protocol:
- DNA Extraction: Lyse the food sample to release DNA. Purify the DNA using a method (e.g., spin-column technology) that removes inhibitors such as polysaccharides, fats, and polyphenols that can compromise the PCR reaction [5].
- Prepare PCR Mix: Combine the purified DNA template with a master mix containing sequence-specific primers (designed to target a unique gene of the allergenic source, such as a mitochondrial or tropomyosin gene), nucleotides (dNTPs), a thermostable DNA polymerase, and a fluorescent dye (e.g., SYBR Green) or probe (e.g., TaqMan) for real-time detection [5] [2].
- Real-Time PCR Amplification: Place the reaction plate in a thermal cycler. The instrument runs through repeated cycles of denaturation (separating DNA strands), annealing (primers bind to target sequence), and elongation (polymerase synthesizes new DNA). The fluorescence is monitored at each cycle [5] [2].
- Data Analysis: The cycle threshold (Ct), at which the fluorescence exceeds a background level, is determined. A lower Ct indicates a higher initial amount of target DNA. Results are interpreted qualitatively (presence/absence) or semi-quantitatively by comparison to standard curves [2] [4].
Research Reagent Solutions and Essential Materials
Selecting the right reagents is critical for the success of any allergen detection assay. The following table lists key materials and their functions.
Table 3: Essential research reagents and materials for ELISA and PCR allergen detection.
| Reagent / Material | Function | Key Considerations for Researchers |
|---|---|---|
| Capture & Detection Antibodies (ELISA) | Specifically bind to the target allergenic protein for capture and signal generation [4]. | Select antibody pairs validated for the specific allergen (e.g., Ara h 1 for peanut). Cross-reactivity must be characterized [18]. |
| Protein Extraction Buffer (ELISA) | Solubilizes proteins from the food matrix while preserving antigenic structure [18]. | Buffer composition is critical, especially for processed foods. Optimized, commercial buffers can significantly improve recovery [18]. |
| Species-Specific Primers (PCR) | Short DNA sequences that define the specific region of the allergen's genome to be amplified [5] [2]. | Primers must be highly specific to avoid cross-reaction with non-target species. Target genes include 12S rRNA (crustaceans) or tropomyosin [2]. |
| DNA Polymerase (PCR) | Enzyme that synthesizes new DNA strands during the amplification process [5]. | Use a thermostable polymerase suitable for qPCR. Enzyme blends designed to tolerate common PCR inhibitors from food matrices are available. |
| Real-Time PCR Master Mix | A pre-mixed solution containing dNTPs, buffers, salts, and a fluorescent reporting system (dye or probe) [42]. | Simplifies reaction setup. Choose between intercalating dye (e.g., SYBR Green) for simplicity or probe-based (e.g., TaqMan) for higher specificity in multiplex assays. |
The choice between ELISA and PCR is a strategic decision that must be matrix-driven. ELISA remains the gold standard for direct, quantitative protein detection in a wide range of foods and is often specified for regulatory compliance, such as gluten testing [19] [3]. However, PCR emerges as a superior tool for qualitative detection in complex, processed matrices where protein integrity is compromised, leveraging the greater stability of DNA [2] [19]. For the most comprehensive risk assessment, an integrated approach using both methods provides the most robust safety net, combining the direct quantification of ELISA with the high sensitivity of PCR for challenging matrices [5].
Future developments in allergen detection are moving towards methods like liquid chromatography-mass spectrometry (LC-MS/MS), which can simultaneously detect and confirm multiple allergenic proteins, even in processed foods where ELISA has failed [41]. Furthermore, the rise of biosensors promises rapid, on-site detection capabilities, which could revolutionize environmental monitoring and supply chain testing [3]. For now, a thorough understanding of the principles, performance, and limitations of both ELISA and PCR, as detailed in this guide, is fundamental for researchers and scientists to make informed decisions that protect consumer health and ensure product safety.
Food allergies represent a significant public health concern, with crustacean shellfish being one of the major allergens regulated by food safety agencies worldwide [2]. For allergic individuals, avoiding foods containing crustaceans is imperative, which necessitates highly sensitive and accurate detection methods to verify labeling and detect potential cross-contamination [44]. Two primary methodologies employed for allergen detection are protein-based ELISA (Enzyme-Linked Immunosorbent Assay) and DNA-based PCR (Polymerase Chain Reaction) [4]. This case study provides a comprehensive comparison of these technologies, with a specific focus on the superior dynamic range of PCR-based methods for detecting crustacean allergens in processed foods, presenting experimental data and protocols to guide researchers and food safety professionals in method selection and implementation.
Methodological Comparison: ELISA vs. PCR
Fundamental Principles and Targets
ELISA and PCR differ fundamentally in their detection targets and operational principles, which directly impacts their performance characteristics.
ELISA (Enzyme-Linked Immunosorbent Assay): This immunoassay detects allergenic proteins directly. The most common format for allergen testing is the sandwich ELISA, which uses two antibodies that bind to different epitopes of the target protein [4]. This dual-antibody system provides high specificity, but its effectiveness can be compromised when food processing alters protein structure, potentially masking antibody binding sites.
PCR (Polymerase Chain Reaction): This molecular technique detects species-specific DNA sequences rather than proteins. By targeting conserved genomic regions, such as mitochondrial genes (12S rRNA, 16S rRNA), PCR amplifies and detects trace amounts of DNA from allergenic ingredients [2] [44]. DNA is generally more stable than proteins during food processing, remaining detectable even when proteins are denatured.
Experimental Workflows
The experimental workflows for these techniques differ significantly, contributing to their distinct performance profiles.
Diagram 1: Comparative workflows for ELISA and PCR methods.
ELISA Protocol for Crustacean Detection
The standard sandwich ELISA protocol for crustacean allergen detection involves these critical steps [4]:
- Protein Extraction: Food samples are homogenized in an appropriate extraction buffer to solubilize proteins. The buffer composition is crucial for efficient extraction and may vary based on the food matrix.
- Antibody Coating: Wells of a microplate are coated with a capture antibody specific to a crustacean allergen protein (e.g., tropomyosin).
- Incubation and Binding: The protein extract is added to the wells. If the target allergen is present, it binds to the capture antibody.
- Detection Antibody: A second enzyme-conjugated antibody, specific to a different epitope on the allergen, is added to form a "sandwich" complex.
- Signal Development: A substrate solution is added. The enzyme converts the substrate to a colored product.
- Quantification: The color intensity, measured spectrophotometrically, is proportional to the allergen concentration in the sample, calculated against a standard curve.
PCR Protocol for Crustacean Detection
A typical real-time PCR (qPCR) protocol for crustacean detection follows this sequence [44] [45]:
- DNA Extraction: DNA is purified from the food sample using commercial kits, often incorporating steps to remove PCR inhibitors common in complex matrices.
- Primer/Probe Design: Primers and probes are designed to target conserved crustacean-specific DNA sequences. Common targets include:
- PCR Amplification: The reaction mix containing template DNA, primers, probes, nucleotides, and polymerase is subjected to thermal cycling (e.g., 40-45 cycles of denaturation, annealing, and extension).
- Real-Time Detection: Fluorescence is monitored during each cycle. The cycle threshold (Ct) at which fluorescence exceeds the background is recorded.
- Quantification: The Ct value is compared to a standard curve from known DNA concentrations to quantify the amount of crustacean DNA in the sample.
Performance Comparison: Quantitative Data
Direct comparative studies reveal significant differences in the performance metrics of ELISA and PCR, particularly regarding dynamic range and sensitivity.
Dynamic Range and Sensitivity
A direct comparative study using identical split samples for crustacean allergen detection demonstrated a stark contrast in dynamic range [2].
Table 1: Comparative Analytical Performance of PCR and ELISA
| Method | Target Molecule | Dynamic Range | Limit of Detection (LOD) | Matrix Interference |
|---|---|---|---|---|
| Real-time PCR | DNA (12S rRNA, 16S rRNA) | 0.1 - 100,000 mg/kg [2] | 0.1 pg shrimp DNA [44] | Minimal interference observed [2] |
| Commercial ELISA | Total crustacean protein / Tropomyosin | 200 - 4,000 mg/kg [2] | Varies by kit (typically ppm level) | Significant interference in complex matrices [2] |
The data shows that PCR offers a broader dynamic range, spanning six orders of magnitude, compared to the more constrained linear range of ELISA. Furthermore, PCR demonstrated minimal matrix interference in complex foods like Manhattan clam chowder and fish sauce, whereas ELISA results were significantly affected by the food matrix [2].
Specificity and Cross-Reactivity
Both methods can achieve high specificity, but their bases differ.
- PCR Specificity: Primers can be designed for different specificity levels. Assays can target specific species (e.g., crab [46]) or broad groups (e.g., all Decapoda crustaceans [45]). A well-designed duplex qPCR system showed no cross-reactivity with over 100 different plant and animal species, including farmed insects [45].
- ELISA Specificity: Dependent on antibody specificity. Cross-reactivity can occur with related proteins from non-target species, potentially causing false positives. For example, an ELISA for shrimp tropomyosin might cross-react with other shellfish tropomyosins [4].
The Researcher's Toolkit: Essential Reagents and Materials
Successful implementation of PCR or ELISA methods requires specific reagents and materials. The following table outlines key solutions for developing these assays.
Table 2: Essential Research Reagent Solutions for Allergen Detection
| Item | Function in ELISA | Function in PCR |
|---|---|---|
| Specific Antibodies | Capture and detection antibodies specific to crustacean allergen proteins (e.g., tropomyosin). | Not applicable. |
| Primers & Probes | Not applicable. | Oligonucleotides targeting conserved crustacean genes (e.g., 12S/16S rRNA). |
| DNA Polymerase | Not applicable. | Enzyme for amplifying target DNA sequences in thermal cycling. |
| Protein Extraction Buffer | Solubilizes and extracts proteins from complex food matrices for ELISA analysis. | Not applicable. |
| DNA Extraction Kit | Not applicable. | Isolates high-quality, inhibitor-free DNA from complex food matrices. |
| Enzyme Substrate | Chromogenic or chemiluminescent substrate for signal generation in ELISA. | Not applicable. |
| qPCR Master Mix | Not applicable. | Optimized buffer containing dNTPs, polymerase, and salts for efficient amplification. |
Discussion and Research Implications
Advantages of PCR's Broader Dynamic Range
The extensive dynamic range of PCR (0.1-100,000 mg/kg) provides several practical advantages for researchers and food safety applications [2]:
- Versatility in Sample Analysis: A single PCR assay can accurately quantify both trace-level cross-contamination and significant allergen amounts in composite foods, eliminating the need for sample dilution or multiple tests.
- Superior Sensitivity for Precautionary Labeling: The high sensitivity (LOD of 0.1 pg DNA) enables detection of allergenic residues at levels far below the threshold that might trigger a reaction in sensitive individuals, providing a robust tool for verifying "free-from" claims and assessing cleaning validation [44] [5].
- Robustness in Processed Foods: DNA's relative stability to heat and processing compared to proteins makes PCR particularly suitable for detecting crustaceans in cooked, baked, or sterilized products where protein epitopes may be denatured [4] [5].
Limitations and Complementary Use of ELISA
Despite its advantages in dynamic range, PCR has limitations. It detects species-specific DNA, not the allergenic protein itself. Therefore, in scenarios where the correlation between DNA copy number and protein allergenicity is uncertain, ELISA, which directly measures the protein, remains the gold standard [4]. Furthermore, for some allergens like gluten or in cases of egg and milk detection, ELISA is the prescribed or more practical method [5].
An integrated approach, using both PCR and ELISA, can offer the most comprehensive solution [5]. PCR can serve as a highly sensitive screening tool, while ELISA can provide confirmation and quantify the actual allergenic protein, especially in complex or ambiguous cases.
This case study demonstrates that PCR offers a distinct advantage over ELISA for the detection of crustacean shellfish allergens, primarily due to its significantly broader dynamic range and superior sensitivity. The ability of PCR-based methods to detect from 0.1 to 100,000 mg/kg, coupled with their resilience to matrix effects and food processing, makes them an powerful tool for food safety research and compliance monitoring [2]. While ELISA continues to be valuable for direct protein quantification, the analytical performance of PCR, particularly when targeting mitochondrial genes, positions it as the preferred methodology for applications requiring the highest level of sensitivity and a wide quantitative range. Researchers should consider these performance characteristics alongside their specific experimental needs, such as the food matrix and required detection threshold, when selecting the most appropriate allergen detection method.
Food allergies represent a significant public health concern worldwide, and for affected individuals, strict avoidance of allergenic foods is the only effective preventive measure [3]. This necessitates highly sensitive and accurate detection methods to ensure compliance with food labeling regulations and to protect consumer health [2]. The Codex Alimentarius Commission, as the international food standards setting body, has established specific guidelines for allergen detection, particularly for gluten [47] [48].
This case study examines the application of Enzyme-Linked Immunosorbent Assay (ELISA) for detecting gluten and milk allergens in alignment with Codex Alimentarius standards, framing this analysis within broader research comparing ELISA with Polymerase Chain Reaction (PCR) methodologies for allergen detection in processed foods. The performance characteristics, experimental protocols, and regulatory standing of each method are critically evaluated to provide researchers and drug development professionals with evidence-based guidance for method selection.
Principles and Mechanisms of Detection
Fundamental ELISA Technology
ELISA is a plate-based assay technique designed for detecting and quantifying soluble substances such as peptides, proteins, antibodies, and hormones [49]. In the context of allergen detection, ELISA operates on the principle of antigen-antibody interaction, using antibodies that specifically bind to allergenic proteins present in food samples [5]. The assay utilizes a microplate format, typically with 96- or 384-well polystyrene plates that passively bind antibodies and proteins [49].
The core steps of an ELISA include:
- Coating/Capture: Direct or indirect immobilization of antigens to the surface of polystyrene microplate wells
- Plate Blocking: Addition of irrelevant protein or other molecules to cover all unsaturated surface-binding sites of the microplate wells
- Probing/Detection: Incubation with antigen-specific antibodies that affinity-bind to the antigens
- Signal Measurement: Detection of the signal generated via direct or secondary tag on the specific antibody [49]
Among various ELISA formats, the sandwich ELISA is predominantly used for allergen detection because it offers high sensitivity and specificity through dual antibody recognition [49] [4]. In this format, the target antigen is bound between two primary antibodies - a capture antibody and a detection antibody - each detecting a different epitope of the antigen [49].
PCR Technology Fundamentals
In contrast to ELISA's protein-based detection, PCR is a molecular biology technique that amplifies specific DNA sequences unique to allergenic species [5]. The process begins with extracting DNA from food samples, which is then mixed with primers that hybridize specifically with the target allergen's DNA [5]. Through repeated cycles of heating and cooling in a thermal cycler, the target DNA sequence is exponentially amplified, with detection typically performed via real-time PCR methods [5].
Diagram 1: Comparative workflows of ELISA and PCR detection methodologies
Codex Alimentarius Standards and Methodological Validation
Gluten Detection Standards
The Codex Alimentarius Commission has established specific guidelines for gluten-free labeling, stating that any food product containing more than 20 mg/kg gluten cannot be considered or labeled as "gluten-free" [48]. For gluten detection, Codex endorses the R5 antibody-based ELISA method in combination with a patented cocktail solution for extraction [47] [48]. This method has been adopted as a Codex Alimentarius Type I method and was further endorsed by the AOAC as Official Method of Analysis First Action number 2012.01 [47].
The R5 antibody specifically targets gliadin, the alcohol-soluble fraction of gluten proteins found in wheat, rye, and barley [47]. The method's reliability stems from its recognition of multiple gliadin fractions (ω, α/β, and γ-gliadins) and its compatibility with a special cocktail extraction procedure developed for heat-processed samples [47] [48].
Milk Allergen Detection Considerations
While Codex Alimentarius does not prescribe a specific detection method for milk allergens, ELISA remains the preferred analytical technique due to its ability to directly detect allergenic milk proteins such as casein [5] [4]. The sandwich ELISA format is particularly suitable for milk allergen detection because milk proteins like casein are large enough to allow simultaneous binding of two antibodies, enabling highly specific and quantitative detection [4].
Experimental Protocols and Research Reagent Solutions
ELISA Protocol for Gluten Detection (Codex Alimentarius Method)
Sample Preparation:
- Weigh 0.5 g of homogenized test sample into a 15 mL centrifuge tube
- Add 5 mL of Cocktail extraction solution (patented solution containing reducing agents)
- Vortex vigorously, then shake for 60 minutes on a horizontal shaker at 22-25°C
- Centrifuge at 4500 × g for 10 minutes
- Dilute supernatant with PBS buffer as needed [47] [50]
Assay Procedure:
- Coat microplate wells with R5 antibody (capture antibody) in carbonate-bicarbonate buffer (pH 9.6) and incubate overnight at 4°C
- Block plates with 5% PBS-milk for 1 hour at 25°C to cover unsaturated binding sites
- Add extracted samples and standards to wells in triplicate, incubate for 2 hours at 25°C
- Wash plates 3 times with PBS-T (PBS with 0.05% Tween-20)
- Add enzyme-conjugated R5 detection antibody diluted in PBS-T with 0.1% BSA, incubate for 1 hour at 25°C
- Wash plates 3 times with PBS-T
- Add p-Nitrophenyl phosphate (pNPP) substrate and incubate for 1 hour at 25°C
- Measure absorbance at 405 nm with reference at 490 nm using a microplate reader [47] [50]
Quantification:
- Generate standard curve using gliadin standards of known concentration
- Calculate gluten content in samples based on standard curve, applying appropriate dilution factors
- Express results as mg/kg (ppm) of gluten [50]
PCR Protocol for Allergen Detection
Sample Preparation:
- Extract DNA from 0.2-0.5 g of test sample using commercial DNA extraction kits
- Quantify DNA concentration and purity using spectrophotometry
- Adjust DNA concentration to working dilution for PCR amplification [5]
Assay Procedure:
- Prepare PCR reaction mix containing:
- 1× PCR buffer
- 2.5 mM MgCl₂
- 200 µM of each dNTP
- 0.2 µM of each forward and reverse primer (species-specific)
- 1.25 U of DNA polymerase
- 5 µL of template DNA
- Perform real-time PCR amplification using the following cycling conditions:
- Initial denaturation: 95°C for 10 minutes
- 40-45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing: 60°C for 30 seconds (primer-specific temperature)
- Extension: 72°C for 30 seconds
- Monitor fluorescence during each cycle for amplicon detection
- Analyze amplification curves and determine Cq (quantification cycle) values [5]
Data Analysis:
- Use standard curve method with DNA standards of known concentration for quantification
- Express results as DNA content, with conversion to allergen content possible using reference materials [47]
Research Reagent Solutions
Table 1: Essential research reagents for allergen detection methodologies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| R5 Monoclonal Antibody | Specifically recognizes gliadin epitopes in wheat, rye, and barley | RIDASCREEN Gliadin, Veratox for Gliadin R5 [47] [50] |
| Cocktail Extraction Solution | Patented solution with reducing agents for efficient gluten extraction from processed foods | Used with RIDASREEN Gliadin per Codex method [47] |
| Anti-Casein Antibodies | Specifically targets milk allergenic proteins for detection | Milk allergen ELISA test kits [4] |
| Species-Specific Primers | DNA sequences that hybridize to unique allergen gene targets | SureFood ALLERGEN Gluten qPCR [47] |
| Microplate Washers | Automated washing of microplate wells between assay steps | Standard ELISA instrumentation [49] |
| Real-Time PCR Instruments | Thermal cycling with fluorescence detection for DNA amplification | All real-time PCR systems [5] |
| Protein Standards | Calibrators of known concentration for quantification | Gliadin standards for calibration curves [50] |
| DNA Standards | Calibrators of known DNA concentration for PCR quantification | Quantard 40 for gluten-containing cereals [47] |
Comparative Performance Data
Quantitative Method Comparison
Table 2: Performance characteristics of ELISA versus PCR for allergen detection
| Parameter | ELISA | PCR |
|---|---|---|
| Detection Target | Proteins (direct detection of allergens) [5] [51] | DNA (indirect indicator of allergenic potential) [5] [51] |
| Sensitivity | High to very high (capable of detecting < 5 mg/kg gluten) [48] | Very high (capable of detecting trace DNA amounts) [5] |
| Dynamic Range | 200-4000 mg/kg (crustacean allergen study) [2] | 0.1-106 mg/kg (crustacean allergen study) [2] |
| Quantification Capability | Directly quantitative (measures allergen concentration) [5] [51] | Qualitative or semi-quantitative (measures DNA, not protein) [5] [51] |
| Matrix Interference | Susceptible to matrix effects in complex foods [2] | Reduced matrix interference in most applications [2] |
| Effect of Food Processing | Protein denaturation may affect detection [3] [5] | DNA degradation in highly processed foods (oils, gelatin) [51] |
| Multiplexing Capability | Limited to single analyte per well [51] | Possible (e.g., 3 parameters simultaneously) [51] |
| Assay Time | 0.5 - 2 hours [51] | 2 - 3 hours [51] |
| Regulatory Status | Codex Alimentarius endorsed for gluten [47] [48] | Official method in some countries (e.g., Germany) [3] |
Matrix-Specific Performance
Research studies demonstrate that the performance of both ELISA and PCR varies significantly across different food matrices. In a comparative study of crustacean shellfish allergen detection, PCR assays demonstrated a broader dynamic range (0.1-106 mg/kg) compared to ELISA (200-4000 mg/kg) and did not show matrix interference in Manhattan clam chowder and fish sauce, unlike ELISA methods [2].
For gluten detection, the R5 ELISA method has been collaboratively studied with special emphasis on incurred samples where gluten proteins were exposed to different food processing conditions, leading to AOAC extension of the method's scope to an "in foods" applicability in 2021 [47]. PCR methods for gluten detection, such as the SureFood ALLERGEN Gluten qPCR, can detect gluten-containing cereals without differentiation, with quantification based on DNA and subsequent conversion using laboratory reference material [47].
Diagram 2: Decision pathway for selecting appropriate allergen detection methodology
Limitations and Complementary Applications
Method-Specific Constraints
ELISA Limitations:
- Epitope Dependency: Sandwich ELISA requires at least two sterically distant epitopes for binding, making it unsuitable for detecting hydrolyzed proteins where epitopes may be destroyed [48]
- Antibody Cross-Reactivity: Potential for cross-reactivity with non-target proteins, leading to false positives [51]
- Processing Effects: Heat processing can alter protein structures, potentially affecting antibody recognition and detection capability [3] [5]
- Matrix Interference: Complex food matrices may interfere with antibody-antigen interactions [2]
PCR Limitations:
- Indirect Detection: PCR detects DNA rather than the allergenic protein itself, creating potential discrepancies between DNA presence and actual allergenicity [51]
- Detection Gaps: Unable to detect allergens from milk and eggs effectively, as the analysis would only detect cow or chicken DNA without specifying the allergenic component [51]
- Processing Limitations: DNA may not survive all food processing methods; highly processed foods like vegetable oils, gelatin, lecithin, or starch contain little or no DNA [51]
- Quantification Challenges: Provides DNA quantification rather than direct protein measurement, requiring conversion factors that may introduce errors [47]
Integrated Methodological Approaches
For comprehensive allergen detection, an integrated approach utilizing both ELISA and PCR methods provides complementary advantages [5]. This is particularly valuable in several scenarios:
Complex Food Matrices: In products with complex matrices or heavily processed ingredients, combining PCR and ELISA can provide comprehensive analysis. PCR targets DNA sequences that may survive processing, while ELISA provides quantitative protein data [5].
Regulatory Compliance and Confirmatory Testing: Using ELISA as the primary method for gluten detection per Codex standards, with PCR as a confirmatory method, enhances the reliability of results, particularly for allergen-free claim verification [5].
Method Validation: ISO 17025 accreditation requires demonstration of method validity for specific matrices, which may involve using both technologies to address their respective limitations and provide comprehensive method validation [4].
This case study demonstrates that both ELISA and PCR methodologies offer distinct advantages and limitations for allergen detection in processed foods. ELISA, particularly the R5 antibody-based method endorsed by Codex Alimentarius for gluten detection, provides direct, quantitative measurement of allergenic proteins with established regulatory standing. PCR offers superior specificity and DNA stability in processed foods but remains an indirect detection method.
For researchers and drug development professionals, method selection should be guided by specific application requirements, including the need for direct protein quantification, regulatory compliance, food matrix complexity, and processing effects. The R5 ELISA method remains the gold standard for gluten detection in compliance with Codex Alimentarius standards, while PCR serves as a valuable complementary technique for specific applications where DNA detection provides advantages. An integrated approach, utilizing both methodologies' complementary strengths, often provides the most comprehensive solution for ensuring food safety and regulatory compliance in allergen detection.
In the fields of diagnostic medicine and food safety, the demand for rapid, on-site screening tools has never been greater. Traditional laboratory methods like Enzyme-Linked Immunosorbent Assays (ELISA) and Polymerase Chain Reaction (PCR), while reliable and sensitive, are time-consuming, require specialized equipment, and must be performed in laboratory settings by trained personnel. Lateral Flow Assays (LFAs) have emerged as a powerful alternative technology that centralizes the aspect of self-evaluation, offering promising potential for rapid management of public health and safety in remote areas and point-of-care settings [52]. These paper-based point-of-care detection platforms have gained paramount approval due to their ease of utility, low cost, and rapid signal readout [52]. Within the specific context of food allergen detection—a critical public health issue where avoidance is the primary preventive measure—LFAs offer a practical solution for manufacturers needing to verify the safety of their products directly in production facilities [53] [3]. This guide provides an objective comparison of LFA performance against established alternatives, supported by experimental data and detailed methodologies relevant to researchers and scientists.
Fundamental Principles and Comparative Assay Mechanisms
How Lateral Flow Assays Work
LFAs are simple devices that use capillary action to move a liquid sample through various zones on a strip. A typical LFA consists of overlapping membranes mounted on a backing card: a sample pad, conjugate pad, detection membrane (usually nitrocellulose), and absorbent pad [54]. The sample is applied to the sample pad, which is impregnated with buffer salts and surfactants to make the sample suitable for interaction with the detection system [54]. The sample then migrates to the conjugate pad, containing antibodies specific to the target analyte conjugated to colored or fluorescent particles (most commonly colloidal gold) [54]. As the sample continues to flow, it enters the detection zone where specific biological components (antibodies or antigens) are immobilized in lines. The recognition of the target analyte produces a visible response on the test line, while a control line indicates proper liquid flow [54].
Two primary formats exist for LFAs: direct (sandwich) and competitive assays. Direct assays are typically used for larger analytes with multiple antigenic sites (such as human chorionic gonadotropin in pregnancy tests), where the presence of a test line indicates a positive result [54]. Competitive assays are employed for small molecules with single antigenic determinants (such as certain food allergens or environmental contaminants), where the absence of a test line indicates a positive result [55]. The competitive format is particularly valuable for small analytes or single epitopes that lack suitable bioreceptor pairs [55].
ELISA (Enzyme-Linked Immunosorbent Assay) is an immunological method performed in laboratory settings using microplates. It involves multiple incubation and washing steps where antigens or antibodies are immobilized on solid surfaces and detected using enzyme-linked conjugates that produce a measurable signal, typically through color change [53]. ELISA can be formatted as sandwich (for larger molecules) or competitive (for smaller molecules) assays, similar to LFA principles but performed in plate wells with more complex processing.
PCR (Polymerase Chain Reaction) is a molecular technique that amplifies specific DNA sequences. Unlike immunochemical methods that detect proteins, PCR identifies the genetic material of allergenic foods, making it particularly useful for detecting allergens in highly processed foods where protein structures may be denatured but DNA remains intact [53] [3].
Table 1: Core Technology Comparison for Allergen Detection
| Parameter | Lateral Flow Assay (LFA) | ELISA | PCR |
|---|---|---|---|
| Target Molecule | Proteins (specific allergens) | Proteins (specific allergens) | DNA (from allergenic ingredients) |
| Detection Principle | Immunochromatography | Immunoassay | Nucleic acid amplification |
| Typical Time to Result | 5-30 minutes [56] | Several hours [53] | 2-4 hours (including DNA extraction) |
| Equipment Needs | Minimal (none for qualitative) | Spectrophotometer, incubators, washers | Thermal cycler, real-time detector |
| Throughput | Low to moderate | High | High |
| Ease of Use | Simple, minimal training | Requires technical expertise | Requires technical expertise |
| Best For | Rapid, on-site screening | Quantitative analysis in lab | Detection in highly processed foods [3] |
Performance Comparison: Experimental Data and Sensitivity Analysis
Direct Performance Studies
Multiple studies have directly compared the performance of LFAs with ELISA and PCR methods. A 2020 study comparing commercial IgG and IgA ELISA with three lateral flow immunoassays for SARS-CoV-2 antibody detection found that ELISA presented better results than LFA, with sensitivities for ELISA anti-SARS-CoV-2 IgG and IgA at 81.5% and 93.1% respectively, with specificities of 100% and 80.6% [57]. The LFI tests showed variable performances, with the best-performing LFA showing overall better results [57].
A more recent 2025 study evaluating an in-house ELISA against a commercial Rapid LFA test (IgG + IgM) found that the in-house ELISA demonstrated a Positive Percent Agreement (PPA) of 83% and Negative Percent Agreement (NPA) of 70.4% compared to the Rapid LFA test [58]. The kappa coefficient between the assays was 0.52 (95% CI 0.46–0.58), indicating only modest agreement between the methods [58].
In food allergen applications, a study on gluten detection demonstrated that LFAs could achieve sensitivity thresholds compliant with regulatory requirements, though ELISA remains the official method adopted by the Codex Alimentarius Commission for gluten allergen testing with a threshold of 20 mg/kg [3].
Table 2: Quantitative Performance Comparison Across Applications
| Application | Method | Sensitivity | Specificity | Detection Limit | Study |
|---|---|---|---|---|---|
| SARS-CoV-2 Antibody Detection | ELISA (IgG) | 81.5% | 100% | N/R | [57] |
| SARS-CoV-2 Antibody Detection | ELISA (IgA) | 93.1% | 80.6% | N/R | [57] |
| SARS-CoV-2 Antibody Detection | LFA (Best Performing) | Variable, lower than ELISA | Variable | N/R | [57] |
| SARS-CoV-2 Antibody Detection | In-house ELISA | PPA: 83% vs LFA | NPA: 70.4% vs LFA | N/R | [58] |
| Food Allergen Detection | ELISA (Official Method) | High | High | 20 mg/kg (gluten) [3] | [3] |
| Food Allergen Detection | PCR | High for DNA-containing allergens | High | Varies by allergen | [53] [3] |
| Food Allergen Detection | LFA | Sufficient for screening | Sufficient for screening | Varies by kit | [53] |
N/R = Not Reported; PPA = Positive Percent Agreement; NPA = Negative Percent Agreement
Key Performance Limitation Considerations
The available comparative data reveals several important trends regarding LFA performance:
Sensitivity Limitations: LFAs generally demonstrate lower sensitivity compared to ELISA and PCR methods, which can lead to false-negative results, particularly when analyte concentrations are low [57] [56].
Specificity Concerns: Cross-reactivity with similar substances can sometimes produce false-positive results in LFAs, though this varies significantly between assays and targets [56].
Quantitative Limitations: Most LFAs provide qualitative (yes/no) results, whereas ELISA offers precise quantification, and PCR can provide semi-quantitative results [53] [56].
Methodological Protocols: Experimental Workflows
Detailed LFA Protocol for Allergen Detection
Principle: The assay uses a competitive format where the target allergen in the sample competes with an immobilized allergen conjugate for binding to labeled antibodies [55].
Materials:
- LFA test strips specific for the target allergen
- Sample extraction solution
- Liquid sample or solid sample homogenate
- Timer
- (Optional) Portable strip reader for semi-quantification
Procedure:
- Sample Preparation: For solid food samples, homogenize 1g of sample with 10mL of extraction buffer. Mix vigorously for 1-2 minutes. Allow large particles to settle or centrifuge briefly.
- Sample Application: Pipette 100-150μL of the extracted supernatant to the sample well of the LFA device.
- Chromatography: Allow the sample to migrate along the strip via capillary action. Wait 10-15 minutes.
- Result Interpretation:
- Positive Result: Only the control line appears, indicating the allergen is present above the detection threshold.
- Negative Result: Both control and test lines appear, indicating the allergen is not detected or is below the detection threshold.
- Invalid Result: No control line appears, regardless of test line visibility.
Comparative ELISA Protocol for Allergen Detection
Principle: Based on a sandwich ELISA format where the target allergen is captured between two specific antibodies [53].
Materials:
- ELISA kit specific for target allergen
- Microplate reader capable of measuring 450nm absorbance
- Micropipettes and tips
- Incubator
- Washing buffer
- Stop solution
Procedure:
- Coating: Add captured antibody to microplate wells and incubate overnight at 4°C.
- Washing: Wash wells 3 times with washing buffer.
- Blocking: Add blocking buffer and incubate 1-2 hours at room temperature.
- Sample Addition: Add standards and samples to appropriate wells, incubate 2 hours at room temperature.
- Washing: Repeat washing step.
- Detection Antibody Addition: Add enzyme-conjugated detection antibody, incubate 2 hours at room temperature.
- Washing: Repeat washing step.
- Substrate Addition: Add enzyme substrate solution, incubate 30 minutes in dark.
- Stop Reaction: Add stop solution.
- Measurement: Read absorbance at 450nm within 30 minutes.
- Calculation: Generate standard curve and calculate allergen concentration in samples.
LFA Workflow Diagram
Advancements in LFA Technology: Enhancing Performance
Strategies for Sensitivity and Specificity Improvement
Recent research has focused on overcoming traditional LFA limitations through various innovative approaches:
Assay Optimization Techniques:
- Flow Rate Control: Adjusting capillary flow rates using physical barriers or membrane treatments can increase interaction times, improving sensitivity. Methods include laser-patterned geometric control barriers (achieving 9.1-fold improvement) [59] and ball pen writing-without-ink (2-fold improvement) [59].
- Immobilization Efficiency: Enhancing bioreceptor attachment to membranes using novel materials like nitrocellulose nanofibers (50-fold improvement) [59] or graphene oxide [59].
Signal Amplification Methods:
- Novel Labels: Evolving from conventional gold nanoparticles to more sensitive labels including:
Recognition Element Enhancement:
- Using novel capture molecules such as nanobodies, short peptides, and aptamers to improve specificity [59].
- Optimizing antibody orientation and density on nanoparticle surfaces [59].
Table 3: Research Reagent Solutions for Enhanced LFA Development
| Reagent Category | Specific Examples | Function | Performance Benefit |
|---|---|---|---|
| Nanoparticle Labels | Colloidal gold, Latex beads, Quantum dots, Carbon nanotubes, SERS tags | Signal generation | Improved visibility and sensitivity [59] [54] |
| Membrane Materials | Nitrocellulose, Nitrocellulose nanofibers, Pillar-based arrays | Platform for bioreceptor immobilization and capillary flow | Better flow control and binding efficiency [59] [54] |
| Recognition Elements | Monoclonal antibodies, Nanobodies, Aptamers, Peptide nucleic acids (PNAs) | Target capture and specificity | Reduced cross-reactivity, better stability [59] |
| Conjugation Chemistries | Streptavidin-biotin, EDC-NHS, Maleimide-thiol | Attachment of recognition elements to labels | Controlled orientation, improved activity [55] |
| Buffer Components | Sucrose, Surfactants, Blocking proteins, Salts | Sample conditioning and conjugate stabilization | Reduced non-specific binding, better flow [54] |
Application in Food Allergen Detection: Specific Considerations
In food allergen testing, each technology has distinct advantages depending on the application context:
LFA Advantages for Allergen Detection:
- Rapid results enable immediate production decisions and environmental monitoring [53]
- Point-of-care utility allows testing in production facilities without sample transport [53]
- Simplicity enables use by quality control staff without specialized laboratory training [53]
- Cost-effectiveness makes frequent monitoring economically feasible [56]
ELISA Advantages:
- Quantification provides precise measurement of allergen levels against regulatory thresholds [53] [3]
- High sensitivity and specificity suitable for compliance testing [3]
- Official status as reference method for certain allergens (e.g., gluten) [3]
PCR Advantages:
- Detection in processed foods where proteins may be denatured but DNA remains detectable [53] [3]
- Specificity for ingredients rather than allergenic proteins specifically [3]
- Multiplexing capability for multiple allergens in a single reaction [3]
Method Selection Decision Tree
Lateral Flow Assays represent a compelling alternative to traditional ELISA and PCR methods for specific applications where speed, portability, and ease of use are prioritized over maximum sensitivity and precise quantification. While ELISA remains the gold standard for quantitative protein detection in laboratory settings, and PCR excels for DNA-based detection in processed foods, LFAs fill the critical niche for on-site screening and rapid decision-making. The performance gap between LFAs and laboratory methods continues to narrow through advancements in nanotechnology, improved recognition elements, and signal amplification strategies. For researchers and food manufacturers, understanding the comparative strengths and limitations of each technology enables appropriate method selection based on specific testing requirements, balancing the need for speed against the necessity for sensitivity in allergen detection protocols.
Overcoming Challenges: Optimization and Troubleshooting for Accurate Results
In the field of food allergen detection, the enzyme-linked immunosorbent assay (ELISA) has long been regarded as the gold standard for routine screening due to its high sensitivity, specificity, and cost-effectiveness [19]. However, a fundamental limitation emerges when analyzing processed foods: protein denaturation. Food processing techniques—including thermal treatment, high-pressure processing, fermentation, and chemical modification—can alter the three-dimensional structure of allergenic proteins [3]. These structural changes may destroy or modify the epitopes that ELISA antibodies are designed to recognize, potentially leading to false-negative results and underestimation of allergen content [51] [3]. This article objectively examines how protein denaturation impacts ELISA performance compared to DNA-based polymerase chain reaction (PCR) methods in processed food matrices, providing researchers with experimental data and methodological considerations for selecting appropriate detection strategies.
Analytical Principles: Differential Targets and Mechanisms
Understanding the core differences between ELISA and PCR methodologies is essential for comprehending their respective limitations and strengths in detecting allergens in processed foods.
ELISA (Protein Detection): This immunological method relies on the specific binding between antibodies and protein epitopes. It directly targets the allergenic proteins themselves, which is advantageous since these proteins directly cause allergic reactions [51]. However, this becomes a limitation when protein structures are altered through processing, as the antibodies may no longer recognize the modified epitopes [3].
PCR (DNA Detection): This molecular technique amplifies species-specific DNA sequences. It indirectly indicates allergen presence by detecting the DNA of the allergenic source [51]. Since DNA is generally more stable than proteins during thermal processing, PCR often maintains detection capability in processed foods where proteins may be denatured [51] [3]. However, a significant limitation is that PCR does not directly detect the allergenic protein itself, potentially leading to false positives if DNA is present without allergenic protein [51].
Table 1: Fundamental Differences Between ELISA and PCR Methodologies
| Characteristic | ELISA | PCR |
|---|---|---|
| Detection Target | Proteins (allergens) [60] | DNA [60] |
| Detection Principle | Antigen-antibody binding [61] | DNA amplification [61] |
| Directness of Measurement | Direct (measures the causative agent) [51] | Indirect (measures associated DNA) [51] |
| Primary Limitation in Processing | Protein denaturation alters epitopes [3] | DNA degradation in extreme processing [51] |
| Quantitative Capability | Directly quantitative [51] | Qualitative or semi-quantitative [51] |
Diagram 1: Analytical pathways for ELISA and PCR in processed foods
Experimental Comparisons: Method Performance in Processed Matrices
Sensitivity Data in Controlled Studies
Comparative studies demonstrate how processing methodologies affect the detection capabilities of both ELISA and PCR techniques. In one study investigating meat species detection in processed products, real-time PCR demonstrated significantly lower detection limits compared to ELISA, particularly for pork detection in binary mixtures where PCR detected 0.10% compared to ELISA's 10.0% detection limit [8]. This hundredfold difference in sensitivity highlights the substantial impact of processing on protein-based detection methods.
For sesame protein detection, a novel ELISA was developed with a limit of detection (LOD) of 0.013 μg/g and limit of quantification (LOQ) of 0.025 μg/g [62]. While these values demonstrate excellent sensitivity for sesame proteins specifically, the recovery rates for incurred food samples (dressing, breads, sauce, and pudding) ranged from 67% to 81%, indicating that matrix effects and processing still impact quantification accuracy even in optimized ELISA systems [62].
Table 2: Comparative Sensitivity Data for ELISA and PCR in Food Analysis
| Study Focus | ELISA Performance | PCR Performance | Food Matrix |
|---|---|---|---|
| Beef Detection [8] | Consistent detection at 1.00% | Consistent detection at 0.50% | Processed meat mixtures |
| Pork Detection [8] | Consistent detection at 10.0% | Consistent detection at 0.10% | Processed meat mixtures |
| Multiple Nuts [63] | Not tested | Detection limit of 0.1 mg/kg | Commercial food products |
| Sesame Protein [62] | LOD: 0.013 μg/g; LOQ: 0.025 μg/g | Not tested | Incurred foods (dressing, bread, sauce) |
Recovery Studies in Processed Foods
Recovery experiments using incurred samples (where allergens are incorporated prior to processing) provide particularly valuable data about method performance. The developed sesame ELISA showed recovery rates between 67% and 81% across various processed food matrices, with the highest recovery in dressings and sauces (81%) and lower recovery in baked goods (67%) [62]. This pattern aligns with the understanding that more intensive thermal processing (like baking) causes greater protein denaturation compared to less intensive processing (like mixing dressings).
PCR methodologies typically demonstrate more consistent recovery across processing types because DNA is less affected by thermal processing, though highly processed foods like vegetable oils, gelatin, lecithin, or starch can present challenges as they contain little or no DNA [51].
Methodological Protocols: Detailed Experimental Approaches
ELISA Protocol for Allergen Detection in Processed Foods
The following protocol is adapted from published methodology for sesame protein detection [62] and can be modified for other allergens:
Sample Extraction Buffer Composition:
- Tris-based neutral buffer solution (20 mM Tris-HCl, pH 7.4)
- 0.1 M sodium sulfite (Na₂SO₃)
- 0.6% (v/v) sodium dodecyl sulfate (SDS)
- 0.1% (w/v) bovine serum albumin (BSA)
- 0.04% (w/v) Tween 20
- 200 mM ethylenediaminetetraacetic acid trisodium salt (EDTA-3Na)
- 0.05% Proclin (preservative)
Extraction Procedure:
- Homogenize 1 g of processed food sample with 20 mL of extraction buffer
- Incubate with continuous shaking for 60 minutes at room temperature
- Centrifuge at 10,000 × g for 15 minutes at 4°C
- Collect supernatant and dilute with sample diluent if necessary
- Filter through 0.45 μm membrane before analysis
Sandwich ELISA Procedure:
- Coat microplate with capture antibody (Clone A, 1 μg/mL in coating buffer) overnight at 4°C
- Block with 1% BSA in PBS for 2 hours at room temperature
- Add extracted samples and standards (50 μL/well), incubate 1 hour at 37°C
- Add detection antibody (Clone B-HRP conjugate, 0.5 μg/mL), incubate 1 hour at 37°C
- Add TMB substrate (100 μL/well), incubate 15-30 minutes in dark
- Stop reaction with 1 N sulfuric acid (50 μL/well)
- Measure absorbance at 450 nm within 30 minutes
Real-Time PCR Protocol for Allergen Detection
This protocol adapts established real-time PCR methodology for nut allergen detection [63]:
DNA Extraction:
- Homogenize 200 mg of processed food sample
- Extract DNA using commercial kit (e.g., DNeasy Mericon Food Kit)
- Determine DNA concentration and purity (A260/A280 ratio)
- Dilute to working concentration (10-50 ng/μL)
Real-Time PCR Reaction Setup:
- Template DNA: 5 μL (25-100 ng total)
- Forward and reverse primers: 0.5 μL each (10 μM stock)
- TaqMan probe: 0.5 μL (10 μM stock)
- 2× TaqMan Master Mix: 12.5 μL
- Nuclease-free water to 25 μL total volume
Thermal Cycling Conditions:
- Initial denaturation: 95°C for 10 minutes
- 45 cycles of:
- Denaturation: 95°C for 15 seconds
- Annealing/Extension: 60°C for 60 seconds
- Data collection during annealing/extension step
Probe Design:
- Target: Plant ITS (Internal Transcribed Spacer) region
- Reporter: 6-carboxyfluorescein (FAM) at 5' end
- Quencher: Blackberry (BBQ) at 3' end
Diagram 2: Experimental workflows for ELISA and PCR protocols
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagents for Allergen Detection Studies
| Reagent/Material | Function | Example Specifications |
|---|---|---|
| Monoclonal Antibodies [62] | Specific recognition of allergen epitopes | Clone A (capture), Clone B (detection); anti-Ses i 6 or Ses i 7 |
| Extraction Buffer with Reducing Agents [62] | Protein extraction from processed matrices | Contains 0.1 M sodium sulfite, 0.6% SDS for protein solubilization |
| TMB Substrate [62] | Colorimetric detection in ELISA | 3,3',5,5'-tetramethylbenzidine, produces blue color upon oxidation |
| TaqMan Probes [63] | Sequence-specific detection in real-time PCR | Dual-labeled with FAM reporter and BBQ quencher |
| DNA Extraction Kit | Isolation of high-quality DNA from food matrices | Commercial kit (e.g., DNeasy Mericon Food Kit) |
| PCR Master Mix | Enzymatic amplification of target DNA | Contains thermostable DNA polymerase, dNTPs, buffer |
| Blocking Agents [62] | Prevent nonspecific binding in ELISA | BSA (1%), casein, or commercial blocking buffers |
| Reference Materials | Method validation and calibration | Certified allergenic ingredients at known concentrations |
The comparative data presented demonstrates that both ELISA and PCR have distinct advantages and limitations for allergen detection in processed foods. ELISA directly measures allergenic proteins but faces significant challenges with epitope denaturation during processing, potentially leading to underestimated allergen content [3]. PCR offers robust detection in processed matrices due to DNA stability but provides only indirect evidence of allergen presence through species identification [51].
For comprehensive allergen risk assessment, a complementary approach utilizing both methodologies provides the most rigorous strategy [19]. ELISA remains the appropriate choice for less processed foods and when direct quantification of allergenic protein is required, while PCR serves as a powerful confirmatory tool for highly processed products where protein denaturation is concern [3] [19]. Researchers should select detection methodologies based on the specific processing conditions of their food matrices and the required detection thresholds, considering that technological advancements continue to enhance both protein-based and DNA-based detection capabilities for improved food safety.
Strategies for Overcoming Matrix Interference in Complex Food Products
The accurate detection of food allergens is a critical public health imperative, directly impacting the safety of allergic consumers. For researchers and scientists in food safety and drug development, the analysis of processed foods presents a significant analytical challenge: matrix interference. Complex food matrices, comprising fats, proteins, carbohydrates, and other compounds, can severely compromise the accuracy of allergen detection methods by interfering with the signal of the target analyte [64]. This interference can lead to both false-positive and false-negative results, carrying substantial risks for consumer health and product compliance. Within this context, the choice between Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR) is pivotal. This guide provides an objective comparison of these two dominant technologies, focusing on their performance in overcoming matrix effects, supported by experimental data and detailed protocols to inform method selection for complex food products.
Fundamental Principles: ELISA vs. PCR
ELISA and PCR employ fundamentally different detection strategies, which dictates their respective vulnerabilities and strengths in the face of matrix interference.
ELISA (Enzyme-Linked Immunosorbent Assay): This method is based on an antigen-antibody interaction to detect specific allergenic proteins. The sandwich ELISA format, which is most common for allergen detection, uses two antibodies that bind to different sites on the target protein. This dual recognition provides high specificity, and the resulting enzymatic color change allows for quantitative measurement of the protein concentration [5] [4]. Since ELISA targets the allergenic protein itself, its performance can be directly affected by food processing, which may denature or alter the protein's structure, thereby changing its reactivity with antibodies.
PCR (Polymerase Chain Reaction): This technique is a molecular biology method that amplifies specific DNA sequences unique to the allergenic source. It does not detect the allergen itself but rather the genetic material of the species from which the allergen originates (e.g., peanut, soy). PCR is renowned for its high sensitivity and is particularly useful when proteins have been denatured during processing, as DNA is generally more stable [5] [4]. However, it is generally considered qualitative or semi-quantitative and cannot distinguish between allergenic and non-allergenic ingredients from the same species.
The following diagram illustrates the core operational difference between these two methods when applied to a complex food matrix.
Comparative Performance Data
The fundamental differences in the targets of ELISA and PCR lead to distinct performance profiles, particularly in the presence of complex matrices and various food processing conditions. The following table summarizes the core attributes of each method based on established use in food safety research.
Table 1: Method Comparison for Allergen Detection in Complex Matrices
| Attribute | ELISA | PCR |
|---|---|---|
| Target Molecule | Allergenic Proteins (e.g., Ara h 1 from peanuts) [4] | Species-Specific DNA (e.g., peanut DNA) [4] |
| Quantitative Output | Fully quantitative (measures protein concentration) [5] [4] | Qualitative or semi-quantitative [5] [4] |
| Impact of Food Processing | Protein denaturation can lead to reduced detection [4] | DNA stability allows for detection in processed foods [5] [4] |
| Susceptibility to Matrix Interference | High (co-extracted compounds can block antibodies) [65] [64] | Moderate (co-extracted inhibitors can impede polymerase) [5] |
| Typical Sensitivity | High (parts per million) [4] | Very High (can detect trace DNA) [4] |
| Best-Suited Matrices | Raw ingredients, less processed foods, dairy, eggs [5] | Heated, fermented, and highly processed foods; complex botanicals [5] [4] |
Experimental data underscores the real-world implications of these attributes. For instance, a study applying targeted LC-MS/MS (a confirmatory technique) to 84 unique food products found significant matrix interference that required specialized software (MADIC) for correction. This research also revealed patterns of allergen contamination, such as milk in chocolate-containing products, where the high-fat matrix is known to pose challenges for immunoassays [65]. Furthermore, the study documented highly variable soy content in foods containing refined ingredients like soybean oil and soy lecithin, a scenario where PCR may be advantageous for confirming the presence of soy, while ELISA is necessary to quantify the potentially allergenic protein [65].
Methodologies for Assessing Matrix Interference
Robust method validation requires experimental assessment of matrix effects. The following protocol, widely used in quantitative LC-MS and adaptable for other techniques, provides a framework for this critical evaluation.
Experimental Protocol: Post-Extraction Addition for Matrix Effect Calculation
This method quantifies the extent to which a matrix suppresses or enhances the analytical signal [64] [66].
Workflow Overview:
Step-by-Step Procedure:
Sample Preparation:
- Prepare a blank sample of the food matrix (e.g., a gluten-free flour, a non-dairy product) using the standard extraction protocol.
- Set A (Solvent Standard): Prepare a known concentration of the target analyte (e.g., a purified allergen protein or a DNA standard) in a neat solvent.
- Set B (Matrix-Matched Standard): Spike the same known concentration of the analyte into the extracted blank matrix sample [64].
Instrumental Analysis: Analyze both sets (A and B) using the intended detection platform (e.g., LC-MS, HPLC, or plate reader for ELISA) under identical conditions. A minimum of five replicates (n=5) is recommended for statistical reliability [64].
Data Analysis and Calculation: Compare the peak areas (or other quantitative signals) from Set A and Set B using the following formula:
Matrix Effect (ME%) = [ (Peak Area B - Peak Area A) / Peak Area A ] × 100% [64]
- Interpretation: An ME% value less than zero indicates signal suppression. A value greater than zero indicates signal enhancement. Best practice guidelines typically recommend implementing mitigation strategies if the absolute value of the matrix effect exceeds 20% [64].
This protocol allows researchers to objectively quantify the interference posed by a specific food matrix and is a critical step in method validation.
Strategies for Matrix Interference Mitigation
Once matrix effects are identified, several strategies can be employed to overcome them.
Table 2: Mitigation Strategies for Matrix Interference
| Strategy | Description | Application Notes |
|---|---|---|
| Sample Clean-Up | Using techniques to remove interfering compounds from the sample extract prior to analysis. | Enhanced Matrix Removal-Lipid (EMR-Lipid) sorbents selectively remove lipids from complex matrices without sacrificing analyte recovery [67]. Solid Phase Extraction (SPE) provides superior matrix clean-up compared to protein precipitation, as demonstrated by a 10-40 fold reduction in matrix load in serum analyses [68]. |
| Analytical Calibration | Using calibration techniques that compensate for the matrix effect. | The Standard Addition Method involves spiking known amounts of analyte into the sample itself, which is effective for endogenous compounds and does not require a blank matrix [66]. Stable Isotope-Labelled Internal Standards (SIL-IS) are the gold standard for LC-MS, as they co-elute with the analyte and correct for ionization suppression, but they are expensive and not always available [66]. |
| Methodological Combination | Using ELISA and PCR in a complementary manner. | In complex or heavily processed ingredients, PCR can first confirm the presence of an allergenic species, after which ELISA can provide quantitative data on the actual protein levels present [5]. This integrated approach leverages the strengths of both techniques. |
The Scientist's Toolkit: Key Research Reagent Solutions
Selecting the appropriate reagents and materials is fundamental to developing robust allergen detection methods. The following table details essential solutions for addressing matrix challenges.
Table 3: Essential Research Reagents for Overcoming Matrix Interference
| Reagent / Material | Function in Mitigating Matrix Interference |
|---|---|
| Enhanced Matrix Removal-Lipid (EMR-Lipid) | A selective sorbent used in a dispersive Solid Phase Extraction (dSPE) format to remove lipids from fatty food extracts without significant loss of target analytes, thereby reducing ionization suppression in MS and background in ELISA [67]. |
| Sandwich ELISA Kits (with validated antibodies) | Commercial kits provide highly specific antibody pairs that bind to different epitopes on the target allergenic protein. This dual recognition reduces cross-reactivity and false positives from matrix components. It is critical to use kits validated for the specific food matrix being tested [5] [4]. |
| Protein Precipitation Reagents (e.g., ACN/Formic Acid) | Solvents like acetonitrile effectively precipitate proteins from the sample, which can be a primary source of interference. This is a simple first clean-up step, though it may offer less comprehensive matrix removal than SPE [68]. |
| Stable Isotope-Labelled Internal Standards (SIL-IS) | The most effective internal standards for mass spectrometry. They are chemically identical to the analyte but have a different mass. They correct for analyte losses during sample preparation and for matrix-induced ionization suppression/enhancement [66]. |
| Species-Specific Primers and Probes | For PCR, these are short, custom DNA sequences designed to bind exclusively to the DNA of the allergenic species. High-quality, specific primers are essential to avoid amplification of non-target DNA from the complex matrix, which leads to false positives [5] [4]. |
The challenge of matrix interference in complex food products necessitates a strategic and informed approach to allergen detection. Neither ELISA nor PCR holds an absolute superiority; instead, their performance is highly context-dependent. ELISA is the method of choice for direct protein quantification in less processed matrices or where regulatory limits are based on protein content. In contrast, PCR offers superior sensitivity and resilience for detecting the presence of an allergenic species in highly processed foods where proteins may be denatured. The most robust food safety strategy, particularly for novel or complex matrices, often involves an integrated approach, using these techniques complementarily. Furthermore, the implementation of rigorous experimental protocols for assessing matrix effects and the judicious application of modern mitigation tools—from advanced sorbents to sophisticated internal standards—are indispensable for generating reliable, accurate data that protects public health.
Optimizing Sample Extraction Protocols for Both Protein and DNA Targets
The accurate detection of food allergens in processed products is fundamentally dependent on the initial sample extraction protocol. Whether targeting proteins for Enzyme-Linked Immunosorbent Assay (ELISA) or DNA for Polymerase Chain Reaction (PCR) analysis, the extraction method directly impacts sensitivity, specificity, and overall assay reliability. For researchers and food safety professionals, selecting and optimizing the appropriate extraction strategy is crucial for validating "free-from" claims, ensuring regulatory compliance, and protecting consumer health [5] [19].
The fundamental challenge lies in the divergent nature of the target molecules: proteins are the actual allergenic components, while DNA serves as a stable marker for the allergenic source. Processing conditions—such as high-temperature baking, fermentation, or extreme pH treatments—can dramatically alter the integrity and extractability of these targets [16]. This article provides a detailed comparative guide to extraction methodologies, supported by experimental data, to empower scientists in making informed decisions for their specific research and quality control applications.
Fundamental Differences Between ELISA and PCR Targets
ELISA and PCR are the two dominant technologies in food allergen detection, but they operate on fundamentally different principles and target different molecules. Understanding this distinction is the first step in optimizing extraction protocols.
ELISA (Enzyme-Linked Immunosorbent Assay): This is an immunoassay that directly detects and quantifies allergenic proteins. It relies on the specific binding of antibodies to protein epitopes. The intensity of the resulting colorimetric signal is proportional to the concentration of the target protein in the sample [5] [69]. The actual protein that triggers an allergic reaction is the analyte.
PCR (Polymerase Chain Reaction): This is a molecular technique that detects the DNA of the allergenic species. It does not detect the allergen itself but rather amplifies specific DNA sequences unique to the allergenic source (e.g., peanut, lupin, hazelnut) to detectable levels [5] [70]. DNA is often more stable than proteins under harsh processing conditions.
The table below summarizes the core differences between these two approaches:
Table 1: Fundamental Comparison of ELISA and PCR Allergen Detection Methods
| Parameter | ELISA | PCR |
|---|---|---|
| Target Molecule | Allergenic proteins (e.g., Ara h 1 in peanut) [4] | Species-specific DNA (e.g., peanut DNA) [4] |
| Detection Principle | Antigen-antibody interaction [5] | Amplification of DNA sequences [5] |
| Primary Output | Quantitative protein concentration [5] [4] | Qualitative or semi-quantitative DNA presence [5] [4] |
| Ideal Use Case | Quantifying intact allergenic protein; regulatory compliance for labels like "gluten-free" [5] [19] | Detecting allergenic ingredients when proteins are denatured; identifying species in complex matrices [5] [16] |
Sample Extraction Protocols for Protein and DNA Targets
Protein Extraction for ELISA-Based Detection
The goal of protein extraction is to solubilize the target allergenic proteins from the food matrix into a liquid buffer while maintaining their immunological reactivity for antibody binding in the ELISA.
Typical Workflow:
- Sample Homogenization: The food sample is finely ground to increase surface area.
- Protein Solubilization: The sample is mixed with an extraction buffer. The composition of this buffer is critical and often proprietary to commercial test kits. It typically contains salts to control ionic strength, detergents to solubilize proteins, and sometimes agents to dissociate protein aggregates.
- Clarification: The mixture is centrifuged to remove insoluble particulate matter, and the supernatant containing the solubilized proteins is collected for analysis [5].
Key Considerations:
- Buffer Composition: The buffer must be optimized to effectively extract the target protein without interfering with the subsequent antibody-binding steps in the ELISA. Cross-reactivity with non-target proteins can be a concern if the antibody specificity is not high [69] [71].
- Matrix Effects: Components like fats, polyphenols, or tannins in the food can co-extract with proteins and interfere with the antibody-antigen reaction, leading to inaccurate results (either false positives or false negatives). Spike-and-recovery tests are essential to validate the protocol for a specific matrix, with ideal recovery rates between 90-110% [71].
DNA Extraction for PCR-Based Detection
DNA extraction aims to obtain a sufficient quantity of pure, amplifiable DNA from the sample. The stability of DNA makes it suitable for detecting allergens in processed foods where proteins may have been denatured.
Standardized Protocol (CTAB Method): A common and robust approach for plant-based foods is the Cetyltrimethyl Ammonium Bromide (CTAB) method, as used in a study detecting lupin traces [70]. The detailed steps are as follows:
- Incubation: 100 mg of sample is incubated with CTAB buffer and proteinase K at 65°C. CTAB is a detergent that helps break down cell walls and membranes and complexes with DNA. Proteinase K degrades nucleases that could destroy DNA.
- RNA Removal: The mixture is treated with RNase A to remove RNA.
- Purification: The solution is extracted with chloroform to remove proteins and other contaminants. After centrifugation, the aqueous phase containing DNA is recovered.
- Precipitation & Washing: DNA is precipitated from the solution using isopropanol or ethanol, then washed with 70% ethanol to remove salts.
- Re-suspension: The final DNA pellet is air-dried and dissolved in a sterile buffer or water [16].
Key Considerations:
- DNA Quality and Purity: The purity and integrity of the extracted DNA are critical for PCR efficiency. Spectrophotometric measurements (A260/A280 and A260/A230 ratios) are used to assess purity and the presence of contaminants like proteins or solvents [16].
- Impact of Processing: Food processing, especially high-temperature treatments, can fragment genomic DNA. For reliable PCR analysis, the target amplicon (the DNA region to be amplified) should be short, typically limited to 200-300 base pairs, to ensure it can be amplified from degraded DNA [16].
The following diagram illustrates the parallel workflows for extracting proteins and DNA from a single food sample, highlighting the divergent paths for ELISA and PCR analysis.
Diagram 1: Parallel Extraction Workflows for Protein and DNA Targets.
Comparative Experimental Data and Performance Analysis
Sensitivity and Detection Limits
The sensitivity of a method defines its lowest reliable detection limit. Experimental data from validation studies provide the best insight into the performance of ELISA and PCR.
Table 2: Comparison of Sensitivity and Detection Limits for Allergen Detection Methods
| Allergen / Matrix | Method | Reported Sensitivity (LOD) | Key Experimental Finding |
|---|---|---|---|
| Lupin (Chocolate cookies, ragù, salad) | Real-time PCR | 0.5 ppm (mg/kg) | The method was specific, robust, and rapid, with a detection limit of 0.5 ppm across complex matrices [70]. |
| Crustacean Shellfish (Manhattan clam chowder, fish sauce) | Real-time PCR | 0.1 - 10⁶ mg/kg | PCR showed a broader dynamic range and no significant matrix interference [2]. |
| Crustacean Shellfish (Manhattan clam chowder, fish sauce) | ELISA | 200 - 4000 mg/kg | The dynamic range was narrower than PCR, and the method was susceptible to matrix interference [2]. |
| Peanut & Tree Nuts (Processed cookie) | Multiplex Real-time PCR | 0.64 mg/kg (approx. 0.1–0.2 mg nut protein/kg) | Using multicopy DNA targets enabled extremely sensitive detection, suitable for protecting highly sensitive consumers [72]. |
| Wheat & Maize Allergens (Baked dough) | PCR | Detected after 60 min at 220°C | DNA targets remained detectable despite high-temperature processing, though with increasing DNA degradation over time [16]. |
Impact of Food Processing on Detection
Food processing is a major factor influencing the choice between ELISA and PCR. A study on wheat and maize allergens investigated the effect of baking temperature and time on the detectability of allergen genes.
- Experimental Protocol: Wheat and maize dough samples were baked at 180°C and 220°C for 10 to 60 minutes. DNA was extracted using the CTAB method, and PCR was performed with primers for specific allergen genes (e.g., HMW-GS for wheat; Zea m 14, Zea m 8, and zein for maize) [16].
- Key Finding: Agarose gel electrophoresis confirmed that genomic DNA degraded with increasing baking temperature and time. However, by designing primers for shorter, stable DNA fragments, the researchers could reliably detect wheat and maize allergens even after 60 minutes of baking at 220°C [16]. This underscores the importance of tailoring the DNA extraction and PCR assay design to the expected level of food processing.
The Scientist's Toolkit: Essential Reagents and Materials
Successful extraction and analysis require specific, high-quality reagents. The following table details key solutions and their functions in the protocols.
Table 3: Key Research Reagent Solutions for Allergen Extraction and Analysis
| Reagent / Solution | Function | Application |
|---|---|---|
| CTAB (Cetyltrimethyl Ammonium Bromide) Buffer | Lyses plant cell walls, complexes with DNA and polysaccharides to purify nucleic acids [16] [72]. | DNA Extraction (PCR) |
| Proteinase K | Broad-spectrum serine protease that degrades nucleases and other contaminating proteins, protecting nucleic acids [16]. | DNA Extraction (PCR) |
| Commercial Protein Extraction Buffer | Typically contains detergents and salts to solubilize proteins while maintaining their native epitope structure for antibody recognition [5] [71]. | Protein Extraction (ELISA) |
| Chloroform | Organic solvent used for liquid-liquid extraction to remove proteins, lipids, and other hydrophobic contaminants from the DNA-containing aqueous phase [16]. | DNA Extraction (PCR) |
| Isopropanol / Ethanol | Water-miscible alcohols used to precipitate nucleic acids out of the aqueous solution for concentration and purification [16]. | DNA Extraction (PCR) |
| Sandwich ELISA Kit | Contains pre-coated capture antibodies, detection antibodies, and enzyme substrates for specific, quantitative detection of allergenic proteins [5] [69]. | Protein Detection & Quantification |
Strategic Selection and Integrated Approach
Choosing between ELISA and PCR is not a matter of which is universally better, but which is more appropriate for the specific analytical question and sample type.
When to Prioritize Protein (ELISA) Extraction:
- When the goal is to quantify the actual allergenic risk, as the immune system reacts to proteins, not DNA [5] [19].
- For compliance with standards that specify protein thresholds (e.g., Codex Alimentarius for gluten) [5].
- When analyzing raw ingredients or minimally processed foods where proteins are likely to be intact and immunoreactive.
When to Prioritize DNA (PCR) Extraction:
- For detecting allergens in highly processed foods (e.g., baked goods, hydrolyzed, or fermented products) where proteins may be denatured or fragmented [5] [16].
- When high sensitivity is required to trace very low levels of contamination [70] [72].
- For species identification in complex matrices with multiple ingredients [5] [4].
An integrated approach, using both methods in combination, can provide the most comprehensive risk assessment. For instance, a Lateral Flow Assay (LFA) can be used for rapid, on-site screening of environmental swabs, followed by confirmatory testing with ELISA or PCR in the laboratory [5]. Similarly, a positive PCR result can be followed by a specific ELISA to determine if the detected allergenic ingredient still presents a protein-level risk. This multi-faceted strategy leverages the complementary strengths of protein and DNA-based detection to enhance overall food safety.
In the field of food safety and diagnostic testing, the accurate detection of allergens is paramount for protecting public health. Two principal methodologies dominate this landscape: the Enzyme-Linked Immunosorbent Assay (ELISA) and the Polymerase Chain Reaction (PCR). ELISA is a biochemical assay that leverages the specificity of antigen-antibody interactions to detect and quantify allergenic proteins, the very molecules that trigger immune responses in susceptible individuals [5]. This method is highly regarded for its specificity, robustness, and quantitative capabilities, making it a standard tool in many laboratories [60]. Conversely, PCR is a molecular biology technique that amplifies specific DNA sequences unique to allergenic sources, enabling the detection of genetic material even in minute quantities [5]. Its exceptional sensitivity makes it particularly valuable for identifying potential allergen contamination when the allergenic protein itself may be degraded or altered [60].
While each method possesses distinct strengths, their individual limitations can compromise result reliability when used in isolation. ELISA's dependence on protein structure can be a drawback in processed foods where high temperatures or fermentation may denature the target proteins, leading to potential false negatives [5]. PCR, while highly sensitive, does not directly detect the allergenic protein itself but rather the presence of source DNA, which could lead to false positives if DNA is detected from a source that no longer contains the functional allergen [4]. Furthermore, PCR is generally considered qualitative or semi-quantitative, making it less suitable for risk assessments that require precise quantification of allergen levels [5] [4]. These methodological constraints highlight the necessity for an integrated approach that synergistically combines both technologies to overcome their individual shortcomings and provide a more comprehensive and reliable assessment of allergen presence.
Fundamental Differences and Performance Comparison
The core distinction between ELISA and PCR lies in their fundamental targets: ELISA detects proteins (the allergens themselves), while PCR detects DNA (the genetic marker of the allergenic source) [60] [4]. This difference dictates their performance characteristics, applications, and susceptibility to interference from food processing. The following table provides a detailed, data-driven comparison of these two methodologies, summarizing their key analytical parameters and performance metrics as established in the scientific literature.
Table 1: Performance Comparison of ELISA and PCR for Allergen Detection
| Parameter | ELISA | PCR |
|---|---|---|
| Target Molecule | Proteins (allergens) [4] | DNA (from allergenic species) [4] |
| Detection Principle | Antigen-Antibody Binding [5] | Nucleic Acid Amplification [9] |
| Sensitivity | High (parts per million) [4] | Very High (can detect trace DNA) [4] |
| Specificity | High, dependent on antibody quality [69] [73] | High, dependent on primer design [9] |
| Quantitative Ability | Fully quantitative [5] [4] | Qualitative or semi-quantitative [5] [4] |
| Effect of Food Processing | Protein denaturation can lead to reduced detection [5] [3] | DNA is more stable; effective for processed foods [5] [3] |
| Risk of False Positives | Cross-reactivity with related proteins [73] | Detection of DNA from non-allergenic tissues or species [4] |
| Risk of False Negatives | If epitopes are damaged during processing | If DNA is degraded or PCR inhibitors are present [9] |
| Time to Result | ~2-3 hours [60] | ~4-5 hours (including DNA extraction) [4] |
| Throughput | High (suitable for batch analysis) [5] | High, with multiplexing capabilities [5] |
| Regulatory Status | Official method for gluten (Codex Alimentarius) [5] [3] | Official method in some countries (e.g., Germany, Japan) [3] |
The data in Table 1 underscores the complementary nature of these two techniques. For instance, ELISA's quantitative strength is counterbalanced by PCR's resilience to processing effects. This complementarity forms the foundational rationale for integrating both methods into a unified analytical workflow to enhance diagnostic accuracy and reliability in complex sample matrices.
Complementary Applications in Processed Food Analysis
The combination of ELISA and PCR becomes particularly powerful when analyzing processed foods, where ingredients undergo physical and chemical transformations that can challenge single-method approaches. In complex or heavily processed food matrices, an integrated strategy leveraging both techniques provides a more reliable safety assessment [5].
ELISA is the preferred method for quantifying specific allergenic proteins in raw materials and less processed foods, making it ideal for validating "free-from" labels and ensuring regulatory compliance for products like milk, eggs, and gluten [5]. For example, it is the standard method for gluten analysis as indicated by the Codex Alimentarius [5]. However, its limitation becomes apparent in baked goods, fermented products, or hydrolyzed proteins, where the protein structure is altered, and antibody binding may be compromised [5].
PCR excels in these challenging scenarios. Since DNA is more thermally stable and survives many manufacturing processes better than proteins, PCR can detect the presence of an allergenic species even when the protein is denatured [5]. This makes it the superior choice for verifying the absence of allergens like celery and fish in processed foods, or for detecting multiple potential contaminants simultaneously through multiplex PCR [5]. A comparative study on malaria detection demonstrated this principle, where PCR successfully identified parasite DNA at early stages, while ELISA, which targets a specific protein, only became effective later in the life cycle [33]. This analogy translates directly to food allergen detection, where PCR can indicate potential contamination that ELISA might miss in highly processed products.
Experimental Workflow for Integrated Allergen Detection
Implementing a combined ELISA and PCR approach requires a systematic workflow that leverages the strengths of each method to verify and complement the other's results. This integrated pathway ensures that the limitations of one technique are counterbalanced by the strengths of the other, leading to a more conclusive and reliable diagnostic outcome. The following diagram visualizes this synergistic experimental workflow.
Integrated ELISA/PCR Workflow for Allergen Detection
The workflow begins with a representative food sample, which is homogenized and divided for parallel analysis. One portion undergoes protein extraction for the ELISA stream (blue nodes), while the other undergoes DNA extraction for the PCR stream (green nodes). The results from both analytical pathways are then integrated and interpreted together (red node), providing a final report with significantly enhanced reliability. This integrated approach is especially critical in situations where results from a single method are ambiguous, or when analyzing novel food matrices with unknown processing effects. For instance, a positive PCR result with a negative ELISA in a baked product might indicate historical contamination or the presence of non-functional protein, while the reverse scenario could suggest cross-reactive proteins in the ELISA [5] [4]. The combined data provides a complete picture that enables researchers and quality control professionals to make more informed decisions about product safety.
PCR-ELISA: A Hybrid Methodology
Beyond running parallel analyses, a more integrated technological solution exists in the form of PCR-ELISA (also known as PCR-ELOSA). This hybrid technique combines the amplification power of PCR with the familiar colorimetric detection of ELISA into a single, streamlined assay [74]. The method fundamentally detects nucleic acids, not proteins, but uses an ELISA-like platform for this detection, offering a unique set of advantages.
The experimental protocol for PCR-ELISA involves three key stages. First, the Amplification stage, where the target DNA is amplified via PCR using nucleotides labeled with a hapten such as digoxigenin (DIG-dUTP), thereby incorporating the label directly into the PCR product [74]. Second, the Immobilization stage, where the denatured, labeled PCR product is hybridized to a biotinylated probe specific to the allergen gene and then captured on a streptavidin-coated microplate via the strong biotin-streptavidin interaction [74]. Finally, the Detection stage, where the immobilized hybrid is detected using an anti-DIG antibody conjugated to an enzyme like peroxidase, followed by the addition of a chromogenic substrate (e.g., ABTS) to produce a measurable color change [74].
Table 2: Comparison of PCR-ELISA with Other Molecular Detection Methods
| Characteristic | Conventional PCR (Gel Electrophoresis) | PCR-ELISA | Real-Time PCR (qPCR) |
|---|---|---|---|
| Detection Method | Agarose gel electrophoresis | Microplate colorimetry | Fluorescence detection in real-time |
| Equipment Needs | Standard lab equipment | Standard lab equipment | Specialized thermal cycler with optical system |
| Quantitative Ability | Not quantitative | Semi-quantitative | Fully quantitative |
| Detection Limit | 1–10 ng/μL [74] | 0.01 ng/μL [74] | 0.25 pg/μL [74] |
| Throughput | Low to moderate | High (amenable to automation) | Moderate |
| Cost per Test | Low | Moderate | High [74] |
| Key Advantage | Simple, low-cost detection | High sensitivity without expensive equipment, semi-quantitative | High sensitivity and full quantification |
| Key Disadvantage | Low sensitivity, not quantitative, uses ethidium bromide | Less sensitive than qPCR | High instrument and reagent cost |
As summarized in Table 2, PCR-ELISA occupies a valuable middle ground. It is more sensitive than conventional PCR with gel detection and provides semi-quantitative data, yet it is more cost-effective than qPCR as it requires standard laboratory equipment rather than a sophisticated fluorescence detection instrument [74]. This makes it a powerful tool for laboratories requiring high-sensitivity, high-throughput screening without the capital investment of a real-time PCR system. Its application has been successfully demonstrated in the detection of various foodborne pathogens like Salmonella and Listeria monocytogenes, as well as in species identification in food products [74].
Essential Research Reagent Solutions
The successful implementation of integrated ELISA and PCR protocols relies on a suite of specialized reagents and tools. The following table details key research solutions required for these advanced allergen detection methodologies.
Table 3: Key Research Reagent Solutions for Integrated Allergen Detection
| Reagent / Tool | Function in Analysis | Application Notes |
|---|---|---|
| High-Affinity Antibodies | Capture and detect specific allergenic proteins in ELISA [69]. | Monoclonal antibodies provide higher specificity; critical for assay sensitivity and reducing cross-reactivity [69] [73]. |
| Biotin-Streptavidin System | Signal amplification in both ELISA and PCR-ELISA [69] [74]. | The 4:1 binding ratio of streptavidin to biotin significantly enhances detection signal [69]. |
| Digoxigenin (DIG)-dUTP | Label for incorporating into PCR amplicons for subsequent detection [74]. | A key component of PCR-ELISA, allowing immunodetection of amplified DNA on a microplate. |
| Protein & DNA Extraction Kits | Isolate target molecules from complex food matrices. | Matrix-specific optimization is crucial for efficiency. Incomplete extraction is a major source of error. |
| Tag DNA Polymerase | Enzyme for amplifying target DNA sequences in PCR [9]. | Thermostable enzyme isolated from Thermus aquaticus; essential for the high-temperature cycling process. |
| Chromogenic Substrates (e.g., ABTS, TMB) | Produce measurable color change in ELISA and PCR-ELISA [74]. | The intensity of the color developed is proportional to the amount of target present. |
| Validated Primer/Probe Sets | Target species-specific DNA sequences for PCR amplification [5]. | Must be designed for unique allergen source genes (e.g., peanut, soy, shellfish) to ensure specificity. |
These reagents form the backbone of reliable allergen testing. Their quality and performance characteristics—such as the affinity of antibodies and the specificity of primers—directly impact the sensitivity, specificity, and overall reliability of both standalone and integrated methods [69] [73]. Researchers must select and validate these reagents rigorously for each specific food matrix to ensure accurate results.
Data-Driven Validation: A Head-to-Head Comparative Analysis of ELISA and PCR
Comparative Analysis of Sensitivity and Specificity in Peer-Reviewed Studies
The accurate detection of food allergens is a critical public health imperative, with undeclared allergens remaining a leading cause of food product recalls worldwide [4]. For researchers, scientists, and drug development professionals, selecting the appropriate analytical method is paramount for ensuring food safety, regulatory compliance, and protecting consumer health. The detection of allergens in processed foods presents unique challenges, as food processing can alter the molecular targets—proteins and DNA—that detection methods rely upon [5].
This guide provides an objective comparison of two principal technologies used in allergen detection: Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR). The performance of these methods is evaluated based on the core diagnostic metrics of sensitivity—the ability to correctly identify true positives—and specificity—the ability to correctly identify true negatives [75]. These metrics are foundational for understanding how a test will perform in real-world scenarios. While sensitivity and specificity are generally considered stable for a given test, predictive values can vary with the prevalence of the allergen in the tested population [75]. The following sections synthesize data from peer-reviewed studies and industry applications to provide a rigorous comparison of ELISA and PCR for allergen detection in processed food research.
Fundamental Principles of ELISA and PCR
The ELISA Method
ELISA is an immunological biochemical assay that detects the presence of allergenic proteins based on antigen-antibody interactions [76]. The test uses enzyme-labelled conjugates and substrates that generate a measurable color change [76]. In the context of food allergen testing, the "sandwich ELISA" format is most common [4]. This format involves two antibodies: a capture antibody bound to a solid phase (typically a microplate well) and a detection antibody that is enzyme-linked. The target protein (allergen) is sandwiched between them. After washing, a substrate is added, and the resulting color intensity, measured spectrophotometrically, is proportional to the amount of allergen present [4] [76]. This method directly detects the allergenic protein itself, which is the molecule that actually elicits an immune response in susceptible individuals.
The PCR Method
PCR is a molecular biology technique that amplifies specific DNA sequences to detect the genetic material of allergenic ingredients [5]. The process involves extracting DNA from a food sample and using primers—short, specific DNA sequences—to target a unique gene region of the allergenic species. Through repeated cycles of heating and cooling in a thermal cycler, the target DNA is exponentially amplified [5] [4]. The amplified DNA is then detected, often in real-time, using fluorescent probes. PCR does not detect the allergenic protein but rather the DNA of the species from which the allergen originates (e.g., peanut, soy, or shellfish) [4]. This is a critical distinction from the ELISA method.
Comparative Performance Data: Sensitivity and Specificity
The performance of diagnostic tests, including those for allergen detection, is most objectively evaluated using the metrics of sensitivity and specificity. Sensitivity measures the proportion of true positives that are correctly identified by the test, while specificity measures the proportion of true negatives that are correctly identified [75]. The table below summarizes the performance characteristics of ELISA and PCR based on data from peer-reviewed studies across various fields, which provide foundational insights into their operational limits.
Table 1: Comparative Performance Metrics of ELISA and PCR
| Metric | ELISA | PCR | Contextual Notes & Sources |
|---|---|---|---|
| Reported Sensitivity | 90.20% [77] | 88.24% [77] | In H. pylori detection, RUT showed highest sensitivity (92.16%) [77]. |
| Reported Specificity | 61.11% - 94.1% [77] [78] | 94.44% [77] | Specificity highly depends on antibody/primers. BP180 ELISA shows 94.1% specificity [78]. |
| Analytical Target | Proteins (the allergen itself) [4] | DNA (from the allergenic species) [4] | Fundamental difference: ELISA detects the culprit molecule, PCR infers its potential presence. |
| Quantification | Fully quantitative [5] [4] | Qualitative or semi-quantitative [4] | ELISA is preferred when concentration (e.g., ppm) is required for compliance [5]. |
| Effect of Processing | Protein structure may be denatured or altered, affecting detection [5] | DNA is more stable and can survive heat, pressure, and pH changes [5] [4] | PCR is often preferred for highly processed, hydrolyzed, or fermented foods [5]. |
The data in Table 1 illustrates that both methods can achieve high sensitivity and specificity, but their performance is highly context-dependent. For instance, a study on Helicobacter pylori detection reported sensitivities of 90.20% for ELISA and 88.24% for PCR, with PCR demonstrating higher specificity (94.44%) than ELISA (61.11%) in that specific context [77]. Furthermore, a meta-analysis of ELISA tests for Bullous Pemphigoid demonstrated that a well-designed assay (BP180 ELISA) can achieve a specificity of 94.1% [78]. This underscores that the quality of reagents, such as the specificity of antibodies in ELISA or primers in PCR, is a major determinant of performance.
Experimental Protocols and Workflows
Detailed ELISA Protocol for Allergen Detection
The sandwich ELISA protocol is a meticulous process that requires precise execution at each stage to ensure reliable and quantitative results [4] [76].
Table 2: Key Research Reagent Solutions for Sandwich ELISA
| Reagent / Equipment | Function | Technical Considerations |
|---|---|---|
| Capture Antibody | Binds specifically to the target allergenic protein and immobilizes it on the microplate. | Defines the foundation of the assay's specificity. Must be high-affinity and well-characterized. |
| Blocking Buffer | Covers unused protein-binding sites on the microplate to prevent non-specific binding. | Typically contains inert proteins (e.g., BSA or casein) to minimize background noise. |
| Detection Antibody | Binds to a different epitope on the captured allergen. Conjugated to an enzyme (e.g., HRP). | Creates the "sandwich." Enzyme conjugation is critical for signal generation. |
| Enzyme Substrate | Converted by the enzyme (e.g., HRP) into a colored product. | Tetramethylbenzidine (TMB) is common, producing a blue color that turns yellow when stopped [76]. |
| Stop Solution | Acidic solution (e.g., H2SO4 or HCl) that halts the enzyme-substrate reaction. | Stabilizes the final color for measurement [76]. |
| ELISA Microplate Reader | Spectrophotometrically measures the optical density (OD) of the solution in each well. | Standard wavelength is 450 nm for TMB. Generates quantitative data based on a standard curve [76]. |
Workflow:
- Coating: The capture antibody, specific to the target allergen, is adsorbed onto the wells of a polystyrene microplate [76].
- Blocking: The plate is treated with a blocking buffer to cover any remaining protein-binding sites, thereby minimizing non-specific binding in subsequent steps [76].
- Sample Addition: The prepared food sample extract is added to the wells. If the target allergen is present, it binds to the capture antibody during incubation.
- Washing: The plate is washed thoroughly with a buffer to remove unbound proteins and other sample components.
- Detection Antibody Addition: The enzyme-conjugated detection antibody is added. It binds to the captured allergen, forming an antibody-allergen-antibody "sandwich."
- Washing: Another wash step removes any unbound detection antibody.
- Substrate Addition: A substrate solution is added. The enzyme linked to the detection antibody catalyzes a reaction that produces a color change.
- Stop and Read: The reaction is stopped with an acid, and the optical density of each well is measured using a microplate reader. The concentration of the allergen is determined by comparing the OD to a standard curve generated from known allergen concentrations [4] [76].
Detailed PCR Protocol for Allergen Detection
The PCR protocol focuses on the extraction and amplification of DNA, providing a highly sensitive method to trace the source of an allergen, even when proteins are degraded.
Workflow:
- DNA Extraction: DNA is isolated and purified from the food sample using standard methods, which may involve lysis, precipitation, and purification steps to remove inhibitors [77] [5].
- Primer Design: Short, specific nucleotide primers are designed to target a unique DNA sequence of the allergenic species (e.g., peanut, celery, or fish) [5] [4].
- PCR Amplification: The extracted DNA is mixed with primers, nucleotides (dNTPs), and a thermostable DNA polymerase (e.g., Taq polymerase) in a thermal cycler [77]. The mixture undergoes repeated cycles of:
- Denaturation: High heat (e.g., 94°C) separates the double-stranded DNA.
- Annealing: Lower temperature allows the primers to bind (hybridize) to their complementary target sequences.
- Extension: The DNA polymerase synthesizes new DNA strands from the primers. These cycles exponentially amplify the target DNA sequence if it is present [77] [5].
- Detection: In real-time PCR (the most common format for allergen testing), the accumulation of amplified DNA is monitored cycle-by-cycle using fluorescent probes. The cycle at which the fluorescence crosses a threshold (Ct value) is related to the initial amount of target DNA, allowing for semi-quantification [5] [4].
Application in Processed Food Research
The choice between ELISA and PCR is heavily influenced by the nature of the food matrix and the degree of processing it has undergone.
ELISA is Best Suited For:
- Quantitative Analysis: When regulatory compliance requires precise measurement of allergen concentration in parts per million (ppm) [5] [4].
- Direct Allergen Measurement: As it detects the protein that causes the allergic reaction, it is the definitive method for assessing potential health risks [4].
- Specific Allergen Targets: It is the standard method for detecting gluten as per the Codex Alimentarius and is preferred for analyzing egg and milk allergens, where PCR may not differentiate between species-specific variants [5].
- Less Processed or Raw Ingredients: Where the protein structures are intact and recognizable by antibodies.
PCR is Best Suited For:
- Highly Processed Foods: For products subjected to high heat, pressure, or extreme pH, where proteins may be denatured and become undetectable by ELISA, but the more stable DNA survives [5] [4].
- Complex Matrices: For foods with multiple ingredients, especially those containing botanical extracts or species where ELISA antibodies might cross-react [5] [4].
- Verification of Allergen-Free Claims: As a highly sensitive screening tool to confirm the absence of DNA from a specific allergenic species [5].
- Specific Challenges: It is particularly useful for detecting allergens like celery (a low-protein matrix) and fish (where no common protein antigen exists for ELISA) [5].
An integrated approach, using both technologies in tandem, often provides the most comprehensive risk assessment. For instance, PCR can be used for initial rapid screening of complex matrices, while ELISA can provide confirmatory, quantitative results on the actual allergenic protein content [5]. This strategy leverages the high sensitivity of PCR with the protein-specific quantification of ELISA.
In the field of processed food allergen detection, selecting the appropriate analytical technique is paramount for ensuring food safety and regulatory compliance. Two of the most prominent methods employed are the Enzyme-Linked Immunosorbent Assay (ELISA) and quantitative Polymerase Chain Reaction (qPCR). While both are powerful tools, they differ fundamentally in what they detect—proteins versus DNA—leading to distinct performance characteristics. Dynamic range, the span of analyte concentrations that can be accurately measured, is a critical parameter that directly impacts a method's applicability. This guide provides an objective comparison of the dynamic range and quantitative precision of ELISA and qPCR, equipping researchers and drug development professionals with the data necessary to select the optimal method for their specific allergen research needs.
Understanding the Core Technologies
ELISA: Immunoassay for Protein Detection
The Enzyme-Linked Immunosorbent Assay (ELISA) is a plate-based assay technique designed for detecting and quantifying soluble substances such as peptides, proteins, antibodies, and hormones [49]. Its principle relies on a highly specific antibody-antigen interaction. The target antigen is immobilized on a solid surface (typically a microplate) and complexed with an antibody linked to a reporter enzyme. Detection is achieved by measuring the enzyme's activity after incubation with a substrate to produce a measurable product, often a color change [49] [76]. The most common format is the sandwich ELISA, which uses two antibodies for high specificity and sensitivity [49].
qPCR: Nucleic Acid Amplification for Gene Detection
Quantitative real-time PCR (qPCR) is a fluorescence-based technique for detecting and quantifying nucleic acids [79]. It monitors the amplification of a target DNA sequence in real-time. The point at which the fluorescence signal exceeds the background level is known as the quantification cycle (Cq), which allows for both qualitative (presence/absence) and quantitative analysis [79]. In the context of allergen detection, qPCR targets specific DNA sequences (genes) unique to the allergenic food source, such as those found in mitochondrial DNA (e.g., 12S rRNA) or specific allergen genes like tropomyosin [2].
Head-to-Head Comparison: Dynamic Range & Quantitative Data
Direct, side-by-side comparisons in scientific studies provide the most reliable performance data. The following table summarizes key findings from a comparative study on crustacean shellfish allergen detection.
Table 1: Experimental Comparison of qPCR and ELISA for Allergen Detection
| Parameter | qPCR (DNA-Based) | ELISA (Protein-Based) | Experimental Context |
|---|---|---|---|
| Dynamic Range | 0.1 - 106 mg/kg [2] | 200 - 4,000 mg/kg [2] | Detection of crustacean allergens in food matrices (Manhattan clam chowder, fish sauce) [2]. |
| Detection Target | Mitochondrial 12S rRNA gene; Tropomyosin gene [2] | Total crustacean protein; Shrimp tropomyosin protein [2] | Four different qPCR methods vs. two commercial ELISA kits on identical split samples [2]. |
| Matrix Interference | No significant interference observed [2] | Demonstrated matrix interference [2] | Matrix effects can compromise ELISA accuracy, whereas PCR was more robust in complex matrices [2]. |
| Qualitative Agreement | Good agreement with ELISA when ELISA was free of matrix interference [2] | Good agreement with PCR in absence of matrix issues [2] | The two methods can corroborate each other, but ELISA's susceptibility to interference is a limitation [2]. |
Interpretation of Comparative Data
The data in Table 1 reveals a stark contrast in dynamic range. qPCR's range spans seven orders of magnitude (from 0.1 mg/kg to 1,000,000 mg/kg), while ELISA's range is confined to a linear range of less than two orders of magnitude (200 to 4,000 mg/kg) [2]. This means qPCR is capable of quantifying allergens across an extremely wide concentration window, from trace contamination to high-level presence, within a single run. ELISA, on the other hand, offers a narrower but highly precise window of quantification, which may require sample dilution to bring concentrations into the assay's optimal range [80].
Experimental Protocols for Allergen Detection
Standard Sandwich ELISA Protocol for Protein Detection
The following workflow details the core steps of a sandwich ELISA, which is commonly used for allergen detection [49].
- Coating: A capture antibody specific to the target allergen is passively adsorbed onto a polystyrene microplate, typically using an alkaline buffer, and incubated for several hours [49].
- Blocking: The plate is treated with an irrelevant protein (e.g., BSA or casein) to cover any remaining hydrophobic binding sites on the plastic, preventing non-specific binding of other components in subsequent steps [49].
- Sample Incubation: The processed food sample extract is added to the well. If the target allergen is present, it binds to the immobilized capture antibody [49] [76].
- Detection Antibody Incubation: An enzyme-conjugated detection antibody, specific to a different epitope on the allergen, is added. This forms the "antibody-antigen-antibody" sandwich [49].
- Substrate Addition: A substrate for the reporter enzyme (e.g., TMB for Horseradish Peroxidase/HRP) is added. The enzyme catalyzes a reaction that produces a colored product [49] [76].
- Signal Measurement & Quantification: The reaction is stopped with an acid, and the intensity of the color is measured spectrophotometrically as Optical Density (OD). The OD is proportional to the amount of allergen in the sample, and its concentration is determined by interpolation from a standard curve [80] [76].
Standard qPCR Protocol for Allergen Gene Detection
The protocol below outlines the process for detecting allergen-specific DNA sequences via a probe-based qPCR assay [2] [81].
- DNA Extraction: Genomic DNA is purified from the processed food sample. This is a critical step, as processing can fragment or degrade DNA [2] [81].
- Reaction Setup: The extracted DNA is combined with a reaction mix containing:
- Amplification & Detection: The qPCR instrument runs thermal cycles (denaturation, annealing, extension). During each cycle, the probe cleaves, separating the reporter from the quencher and increasing fluorescence. This fluorescence is monitored in "real-time" [79] [81].
- Data Analysis: The instrument generates an amplification plot. The Cq (Quantification Cycle), the cycle at which fluorescence crosses a predetermined threshold, is determined. The Cq is inversely proportional to the starting quantity of the target DNA. Absolute or relative quantification is achieved by comparing Cq values to a standard curve of known DNA concentrations [79] [2].
The Scientist's Toolkit: Key Research Reagents & Materials
Successful execution of ELISA and qPCR experiments requires specific, high-quality reagents. The following table itemizes these essential components.
Table 2: Essential Reagents and Materials for Allergen Detection Assays
| Category | Item | Function in the Assay |
|---|---|---|
| ELISA (Protein Detection) | Capture & Detection Antibodies | High-affinity, allergen-specific antibody pair forms the basis of sandwich ELISA specificity [49] [82]. |
| Microplate | 96-well polystyrene plates with high protein-binding capacity and low well-to-well variation [49]. | |
| Enzyme Conjugate | Reporter enzyme (e.g., HRP) linked to the detection antibody for generating signal [49] [76]. | |
| Chromogenic Substrate | Substance (e.g., TMB) enzymatically converted to a colored product for detection [76]. | |
| Blocking Buffer | Protein-based solution (e.g., BSA) used to prevent non-specific antibody binding [49]. | |
| qPCR (DNA Detection) | Species-Specific Primers & Probe | Oligonucleotides designed to uniquely amplify a target gene from the allergenic species [2]. |
| DNA Polymerase | Thermostable enzyme that catalyzes the amplification of the target DNA sequence [79]. | |
| Fluorescent Dye or Labeled Probe | Provides the fluorescence signal for real-time detection of amplified DNA (e.g., SYBR Green, TaqMan probe) [79]. | |
| Nucleic Acid Extraction Kit | For isolating high-quality, inhibitor-free genomic DNA from complex food matrices [2]. | |
| Standard Reference DNA | DNA of known concentration for constructing the standard curve for absolute quantification [79]. |
Strategic Guidance for Method Selection
Choosing between qPCR and ELISA depends on the specific research question and the nature of the sample.
Opt for qPCR for its Broad Dynamic Range and Specificity: qPCR is the superior choice when you need to detect allergens over an extremely wide concentration range, from very low (0.1 mg/kg) to very high levels [2]. It is also highly specific, capable of distinguishing between closely related species based on genetic differences, and is less susceptible to interference from complex food matrices [2].
Choose ELISA for Direct Protein Quantification and Established Use: ELISA is the appropriate method when the goal is to directly measure the allergenic protein itself, which is the molecule that actually elicits an immune response [2]. It is a well-established, standardized technique that is often faster and requires less specialized equipment than qPCR, making it suitable for high-throughput screening in quality control labs [76].
A strategic approach for comprehensive allergen analysis in processed foods could involve using qPCR as a highly sensitive screening tool to detect the presence of any allergenic material, followed by ELISA for precise quantification of the specific allergenic protein in samples that test positive. This two-tiered approach leverages the respective strengths of each technology to provide a robust and reliable assessment of allergen content.
Evaluating Cost, Throughput, and Time-to-Result for Laboratory Efficiency
Food allergy is a significant public health concern worldwide, and for susceptible individuals, the most effective preventive measure is strict avoidance of allergenic foods. This reliance makes accurate food allergen detection not just a scientific endeavor but a critical consumer safety requirement. Within this field, Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR) have emerged as two of the most prominent analytical techniques. This guide provides an objective comparison of ELISA and PCR, focusing on the critical parameters that dictate laboratory efficiency: cost, throughput, and time-to-result. The analysis is framed within the context of detecting allergens in processed foods, where factors like protein denaturation and DNA stability can significantly influence method performance. The decision between these methods is multifaceted, and this guide aims to equip researchers and scientists with the data necessary to select the optimal technology for their specific application.
The fundamental difference between ELISA and PCR lies in their target analyte: ELISA detects proteins, while PCR detects DNA. This distinction is the primary driver for their respective advantages and limitations.
- ELISA (Enzyme-Linked Immunosorbent Assay): This is an immunological method that relies on the specific binding of an antibody to the allergenic protein itself. The detection is achieved through an enzyme-linked conjugate that produces a measurable signal, typically a color change, whose intensity is proportional to the amount of allergen present [5]. As it directly measures the allergenic moiety, it is the officially adopted method for gluten detection by the Codex Alimentarius [3] [5].
- PCR (Polymerase Chain Reaction): This is a molecular biology technique that amplifies specific DNA sequences unique to the allergenic source. Real-time PCR allows for the detection of these amplified DNA sequences, providing a highly sensitive identification of the biological species, even at trace levels [8]. It is an indirect method, as the presence of DNA does not always correlate perfectly with the presence of the intact allergenic protein, but it is exceptionally robust in processed foods where proteins may be denatured [5].
Table 1: Core Principle Comparison of ELISA and PCR.
| Feature | ELISA | PCR |
|---|---|---|
| Target Analyte | Proteins (allergens themselves) [60] [5] | DNA (from the allergenic species) [60] [5] |
| Detection Principle | Antigen-Antibody Binding [5] | Amplification of Specific DNA Sequences [5] |
| Primary Output | Quantification of protein [5] | Qualitative or semi-quantitative detection of species DNA [74] [5] |
Diagram 1: Core Workflow of ELISA vs. PCR.
Comparative Performance Data
Selecting between ELISA and PCR requires a nuanced understanding of their performance across several metrics. The following data, synthesized from comparative studies, provides a foundation for this decision.
Sensitivity and Specificity
Sensitivity, or the lowest detectable amount of an allergen, is a paramount factor for protecting consumer health. A direct comparison of the two methods for species detection in meat products, a common authenticity and allergen concern, clearly demonstrates their differing capabilities.
Table 2: Sensitivity Comparison for Meat Species Detection in Binary Mixtures [8].
| Method | Pork Detection Limit | Beef Detection Limit |
|---|---|---|
| Real-time PCR | 0.10% | 0.50% |
| ELISA | 10.0% | 1.00% |
The data in Table 2 shows that PCR can be significantly more sensitive, detecting pork at a level 100 times lower than ELISA in this specific experimental context. Both methods, however, can achieve 100% specificity when optimized, meaning they do not cross-react with non-target species [8]. For allergens like celery or fish, or in highly processed foods where proteins are degraded, PCR is often the more reliable choice due to the greater stability of DNA [5]. Conversely, ELISA is the definitive method for quantifying specific allergenic proteins like gluten or for differentiating between egg and milk from different animal species [5].
Cost, Time, and Operational Factors
Laboratory efficiency is not solely about performance but also about practicality and cost-effectiveness.
Table 3: Operational and Economic Comparison of ELISA and PCR.
| Factor | ELISA | PCR |
|---|---|---|
| Cost per Test | Generally more cost-effective [19] | More expensive; requires costly reagents and equipment [60] |
| Equipment Cost | Lower; requires standard lab equipment (spectrophotometer) [74] | Higher; requires specialized thermal cyclers (real-time PCR machine) [60] |
| Hands-on Time | Less time-consuming and easier to perform [8] | Requires more technical expertise and stringent sample prep [60] |
| Total Time-to-Result | Typically a few hours [60] | Can be longer due to DNA extraction and amplification [60] |
| Throughput | High; suitable for screening large sample numbers [60] | High, with multiplexing capability for several allergens [5] |
| Ease of Use | User-friendly, established protocol [8] | More complex, requires trained personnel [60] |
Economic models from clinical diagnostics, such as Hepatitis C virus screening, reinforce that a two-step strategy (using a cheaper test like ELISA first) is significantly less costly than using PCR alone in low-prevalence scenarios [83] [84]. This principle is directly transferable to food allergen testing, where ELISA serves as an excellent, cost-effective screening tool.
Experimental Protocols for Allergen Detection
To ensure reproducibility and provide clarity on how comparative data is generated, this section outlines standard protocols for detecting an allergen in a processed food product using both techniques.
ELISA-Based Protocol for Allergen Quantification
Application: Quantitative detection of a specific allergenic protein (e.g., peanut, milk, or gluten) in a processed food matrix [3] [19].
Workflow:
- Sample Homogenization: The food sample is finely ground to ensure a representative aliquot.
- Protein Extraction: Proteins are extracted from a weighed sample portion using an appropriate extraction buffer. The buffer may contain salts and detergents to solubilize proteins and minimize interference from the food matrix. The extract is then centrifuged or filtered to clarify.
- Microplate Incubation: An aliquot of the protein extract is added to the wells of a microplate that are pre-coated with capture antibodies specific to the target allergen. The plate is incubated to allow the allergen to bind to the antibodies.
- Washing: The plate is washed to remove unbound proteins and other matrix components.
- Detection Antibody Incubation: A second antibody, specific to a different epitope on the target allergen and conjugated to an enzyme (e.g., horseradish peroxidase), is added. This forms an antibody-allergen-antibody "sandwich."
- Washing: Another wash step removes unbound detection antibody.
- Substrate Addition: A enzyme substrate is added. The enzyme catalyzes a reaction that produces a colored product.
- Signal Measurement & Quantification: The reaction is stopped, and the absorbance of the solution is measured with a spectrophotometer. The absorbance is compared to a standard curve generated from known allergen concentrations to quantify the allergen in the original sample [5].
PCR-Based Protocol for Allergen Identification
Application: Qualitative or semi-quantitative detection of allergenic species (e.g., peanut, celery, or mustard) in complex or highly processed foods [3] [5].
Workflow:
- Sample Homogenization: The food sample is finely ground.
- DNA Extraction: DNA is purified from a weighed sample portion using a commercial kit. This involves cell lysis, removal of proteins and other contaminants, and DNA isolation. The quality and quantity of the extracted DNA are often assessed.
- Real-Time PCR Setup: The extracted DNA is mixed with primers (short sequences that flank a DNA region unique to the allergenic species), a fluorescently labeled probe (e.g., TaqMan), and a PCR master mix containing DNA polymerase and nucleotides.
- Amplification and Detection: The mixture is placed in a real-time PCR thermal cycler. The instrument runs through cycles of heating and cooling to denature DNA, anneal primers, and extend new DNA strands. The fluorescent signal is monitored in real-time. As the target DNA sequence is amplified, the reporter dye is released from the probe, leading to an increase in fluorescence.
- Data Analysis: The software calculates a cycle threshold (Ct) value, the cycle number at which the fluorescence crosses a defined threshold. A sample is considered positive if its Ct value is below a predetermined level established with control samples. The Ct value can also be used for semi-quantitative estimation [8] [5].
Diagram 2: Experimental Protocol Workflows.
The Scientist's Toolkit: Essential Research Reagent Solutions
The successful implementation of ELISA and PCR protocols depends on a suite of essential reagents and materials. The following table details key components and their functions.
Table 4: Essential Reagents and Materials for Allergen Detection.
| Item | Function | Key Considerations |
|---|---|---|
| Capture & Detection Antibodies (ELISA) | Specifically bind to the target allergenic protein, forming the core of the "sandwich" assay. | Specificity and affinity are critical; cross-reactivity can lead to false positives [3]. |
| Protein Extraction Buffer | Solubilizes and extracts proteins from the complex food matrix while maintaining their immunological identity. | Composition is key for efficient extraction and minimizing interference [5]. |
| DNA Extraction Kit | Purifies and isolates DNA from the sample, removing PCR inhibitors like fats, polyphenols, and proteins. | Yield and purity of DNA directly impact PCR sensitivity and reliability [5]. |
| Primers and Probes (PCR) | Short nucleotide sequences that specifically hybridize to and amplify a unique DNA marker of the allergenic species. | Specificity is paramount; must be validated against a panel of related and unrelated species [8]. |
| Taq DNA Polymerase | The enzyme that synthesizes new DNA strands during the PCR amplification process. | Thermal stability and fidelity are essential for efficient and accurate amplification [74]. |
| dNTPs | The building blocks (nucleotides) used by the polymerase to construct new DNA strands. | Quality and concentration are crucial for optimal amplification efficiency. |
| Microtiter Plates (ELISA) | The solid phase to which the capture antibodies are immobilized, enabling high-throughput processing. | Well-to-well consistency is important for assay precision. |
| Real-Time PCR Plates | Specialized plates compatible with thermal cyclers, designed for optimal heat transfer and signal detection. | Sealing is critical to prevent evaporation and cross-contamination. |
Application Scenarios and Decision Framework
The choice between ELISA and PCR is not universally fixed but depends on the specific analytical question, the nature of the sample, and the required outcome. A hybrid approach often delivers the most robust results.
- When to Prefer ELISA: This is the preferred method for routine quantitative analysis where the target protein is expected to be intact, such as in quality control for gluten-free products [3] [19]. It is also indispensable for verifying claims like "low milk protein" and is the official method for compliance with standards set by the Codex Alimentarius for gluten [5].
- When to Prefer PCR: This method excels in detecting allergens in highly processed foods (e.g., baked, hydrolyzed, or fermented products) where high temperatures or enzymatic treatments may denature proteins, making them undetectable by ELISA [5]. It is also ideal for complex matrices where specific protein antibodies are unavailable or suffer from cross-reactivity, such as with celery and fish allergens [5]. PCR is also superior for qualitative screening of multiple allergens simultaneously through multiplexing.
- Integrated Approach: For the highest level of assurance, particularly with challenging samples, a combination of both methods is advantageous. PCR can first be used as a highly sensitive screening tool to identify the presence of an allergenic species. Subsequently, ELISA can be employed on the positive samples to quantify the amount of intact allergenic protein, providing a complete risk assessment [5].
Diagram 3: Method Selection Decision Framework.
The comparison between ELISA and PCR for allergen detection reveals that neither method is universally superior. Instead, they serve as complementary tools in the food safety arsenal. ELISA stands out for its quantitative power, cost-effectiveness, and direct measurement of the allergenic protein, making it the gold standard for routine compliance and monitoring. PCR offers superior sensitivity and robustness in complex, processed matrices, providing a critical solution for situations where protein-based methods fall short. The most efficient laboratory strategy is one that aligns the technique with the specific application, considering the food matrix, the information required, and economic constraints. As the field evolves, emerging technologies like biosensors and mass spectrometry are gaining traction, but a thorough understanding of ELISA and PCR remains the foundational knowledge for researchers and scientists dedicated to ensuring food safety and protecting public health.
In analytical sciences, particularly when comparing diagnostic methods like ELISA and PCR for allergen detection, establishing the degree of agreement between methods is fundamental to validation. Interrater reliability (IRR) quantifies this agreement, representing the extent to which different methods or raters yield consistent results when evaluating the same samples [85]. While simple percent agreement calculations provide an initial assessment, they fail to account for agreements occurring by chance alone, potentially leading to overly optimistic estimates of methodological consistency [85] [86].
The kappa statistic (κ) addresses this limitation by measuring agreement between categorical classifications while correcting for chance-expected agreement [87] [88]. Developed by Jacob Cohen in 1960, kappa has become an essential metric in method comparison studies across healthcare, biomedical research, and food safety testing [85] [88]. The statistic is particularly valuable when comparing newer methods to established standards, as it provides a robust, chance-corrected measure of classification consistency that helps researchers determine whether methods can be used interchangeably [89] [90].
For researchers comparing ELISA and PCR for allergen detection, understanding kappa statistics is crucial. These methods may classify samples differently based on their detection principles—ELISA detecting proteins and PCR detecting DNA sequences. Kappa provides a standardized framework for quantifying their classification agreement, informing decisions about method selection, validation, and implementation in quality control laboratories.
Fundamental Concepts of Kappa Statistics
Mathematical Foundation
Cohen's kappa coefficient is calculated using the formula:
κ = (p₀ - pₑ) / (1 - pₑ)
where p₀ represents the observed proportion of agreement between methods, and pₑ represents the expected proportion of agreement due to chance [87] [88] [91]. The numerator (p₀ - pₑ) thus represents the agreement beyond what would be expected randomly, while the denominator (1 - pₑ) represents the maximum possible agreement beyond chance [88]. This mathematical formulation produces a statistic that ranges from -1 to +1, with negative values indicating agreement worse than chance, zero indicating agreement equivalent to chance, and positive values indicating agreement better than chance [88].
The calculation of pₑ, the expected chance agreement, depends on the marginal distributions of the ratings. For two raters and categorical data, pₑ is computed as the sum of the products of the corresponding row and column marginal proportions [88]. This calculation assumes that ratings are independent, meaning that the classifications by one method do not influence the classifications by the other method—a key assumption in method comparison studies.
Types of Kappa Statistics
Different variants of kappa have been developed to address various methodological scenarios:
- Cohen's Kappa: Appropriate for two raters (or methods) classifying items into nominal (unordered) categories [88] [90]. This is the most fundamental form of kappa.
- Fleiss' Kappa: Extends Cohen's kappa to accommodate three or more raters or methods [85] [89].
- Weighted Kappa: Used for ordinal data where categories have a natural order (e.g., "low," "medium," "high") [90] [92]. Weighted kappa incorporates differential weighting of disagreements based on their severity, with two primary variants:
Table 1: Comparison of Kappa Statistic Types
| Type | Appropriate Use Cases | Number of Raters/Methods | Data Structure |
|---|---|---|---|
| Cohen's Kappa | Method comparison studies | Two | Nominal categories |
| Fleiss' Kappa | Multi-center studies | Three or more | Nominal categories |
| Linear Weighted Kappa | Ordinal assessments with equally spaced categories | Two | Ordered categories |
| Quadratic Weighted Kappa | Ordinal assessments where major discrepancies are significantly more important | Two | Ordered categories |
For allergen detection studies comparing ELISA and PCR, Cohen's kappa is typically appropriate when classifications are binary (e.g., "detected"/"not detected") or multinomial without natural ordering. If allergen levels are classified ordinally (e.g., "low," "medium," "high" concentration), weighted kappa becomes more appropriate.
Calculation and Interpretation of Kappa
Step-by-Step Calculation
The calculation of Cohen's kappa follows a systematic process that can be illustrated through an example comparing ELISA and PCR for allergen detection:
Step 1: Construct a contingency table comparing results from both methods. For example, when classifying 100 processed food samples for allergen presence:
Table 2: Hypothetical ELISA vs. PCR Comparison for Allergen Detection
| ELISA/PCR | Positive | Negative | Total |
|---|---|---|---|
| Positive | 45 | 10 | 55 |
| Negative | 5 | 40 | 45 |
| Total | 50 | 50 | 100 |
Step 2: Calculate observed agreement (p₀): p₀ = (number of agreements)/(total samples) = (45 + 40)/100 = 0.85 [87]
Step 3: Calculate expected agreement by chance (pₑ):
- Probability both methods randomly say "Positive": (55/100) × (50/100) = 0.275 [88]
- Probability both methods randomly say "Negative": (45/100) × (50/100) = 0.225 [88]
- pₑ = 0.275 + 0.225 = 0.50 [87] [88]
Step 4: Compute kappa: κ = (p₀ - pₑ)/(1 - pₑ) = (0.85 - 0.50)/(1 - 0.50) = 0.35/0.50 = 0.70 [87]
This calculation reveals a kappa of 0.70, indicating substantial agreement beyond chance between the two allergen detection methods.
Interpretation Guidelines
Several frameworks exist for interpreting kappa values, with the most commonly cited being Landis and Koch's benchmarks [88] [91]:
Table 3: Kappa Interpretation Guidelines According to Landis & Koch (1977)
| Kappa Value | Level of Agreement |
|---|---|
| < 0.00 | Poor |
| 0.00 - 0.20 | Slight |
| 0.21 - 0.40 | Fair |
| 0.41 - 0.60 | Moderate |
| 0.61 - 0.80 | Substantial |
| 0.81 - 1.00 | Almost Perfect |
However, interpretation should be context-dependent. The AIAG (Automotive Industry Action Group) suggests that a kappa value of at least 0.75 indicates good agreement for industrial applications, with values above 0.90 preferred for critical assessments [89]. In medical and food safety contexts, where method reliability directly impacts health outcomes, more stringent thresholds are often appropriate.
Several factors influence kappa values beyond the true agreement between methods. Prevalence bias occurs when the distribution of categories is skewed, potentially depressing kappa values [88] [90]. Bias between raters (systematic differences in how categories are used) also affects kappa, with greater impact when agreement is low [88]. The number of categories influences kappa, with more categories typically resulting in lower kappa values [88].
Kappa in Experimental Design: Application to Allergen Detection
Experimental Protocol for Method Comparison
When designing experiments to compare ELISA and PCR for allergen detection in processed foods, the following protocol ensures reliable kappa calculation:
Sample Selection and Preparation:
- Select a representative set of processed food samples (minimum n=50 recommended)
- Include samples with varying allergen concentrations and food matrices
- Ensure proper blinding so technicians performing each method are unaware of the other method's results
- Process samples according to standardized extraction protocols for each method
Data Collection:
- Have trained technicians perform both ELISA and PCR analyses on all samples
- Use established cutoff values for positive/negative classification for each method
- For ordinal classifications, establish clear criteria for concentration categories (e.g., low: 1-5 ppm, medium: 5-20 ppm, high: >20 ppm)
- Record all results in a standardized format for analysis
Data Analysis:
- Construct a contingency table comparing method classifications
- Calculate kappa statistic and its standard error
- Compute confidence intervals (typically 95%) for kappa
- Consider prevalence and bias indices to understand potential limitations
This experimental approach controls for key variables that might affect agreement, ensuring that the calculated kappa accurately reflects methodological consistency rather than experimental artifact.
Visualizing Kappa Calculation
The following diagram illustrates the experimental workflow for comparing allergen detection methods and calculating kappa statistics:
Statistical Relationships
The relationship between kappa, observed agreement, and chance agreement can be visualized as follows:
Comparative Analysis of Agreement Measures
Kappa vs. Other Reliability Measures
While kappa is widely used for categorical data, other reliability measures serve different purposes in method comparison studies:
Table 4: Comparison of Agreement Measures for Method Comparison
| Measure | Data Type | Accounts for Chance | Key Strengths | Key Limitations |
|---|---|---|---|---|
| Percent Agreement | Categorical | No | Intuitive interpretation | Overestimates agreement |
| Cohen's Kappa | Nominal categorical | Yes | Standard for binary classifications | Sensitive to prevalence distribution |
| Weighted Kappa | Ordinal categorical | Yes | Respects ordering of categories | Weighting scheme choice affects results |
| Intraclass Correlation (ICC) | Continuous | Yes | Handles multiple raters/methods | Requires interval-level data |
| Pearson Correlation | Continuous | No | Simple calculation | Does not measure agreement |
For binary classifications in allergen detection (e.g., positive/negative), Cohen's kappa is generally preferred over simple percent agreement due to its correction for chance. When comparing quantitative results (e.g., allergen concentrations), intraclass correlation coefficients (ICC) may be more appropriate, though these require continuous data rather than categorical classifications [86].
Kappa in Published Research
Research by Liu et al. highlighted the importance of appropriate kappa selection in method comparison studies. When comparing radiological assessments, they initially reported a kappa of 0.48, interpreted as "moderate agreement" [90]. However, subsequent analysis revealed that weighted kappa was more appropriate for their ordinal data, yielding values of 0.38 (linear) and 0.40 (quadratic), indicating only "fair" agreement [90]. This case underscores how methodological choices in kappa selection can substantially impact conclusions about method agreement.
In diagnostic method comparisons, kappa values above 0.60 are generally considered minimally acceptable, with values above 0.80 representing strong agreement [89] [86]. For critical applications like allergen detection in foods, where false negatives pose health risks, many researchers advocate for more stringent thresholds (κ > 0.80) to ensure methodological reliability [89].
Practical Considerations and Limitations
Factors Affecting Kappa Values
Several methodological factors influence kappa values in method comparison studies:
- Prevalence Effect: Kappa tends to be lower when the prevalence of a category is very high or very low [88] [90]. In allergen detection, if positive samples are rare (low prevalence), kappa may underestimate true agreement.
- Bias Effect: Systematic differences in how methods assign categories (bias) affects kappa values [88]. For example, if ELISA consistently detects allergens at lower thresholds than PCR, this bias will impact agreement measures.
- Number of Categories: Increasing the number of categories typically reduces kappa values, as chance agreement decreases with more categories [88].
- Sample Size: Small sample sizes lead to less precise kappa estimates with wider confidence intervals [89].
Researchers should report these contextual factors alongside kappa values to facilitate proper interpretation. When prevalence is extreme, additional measures like prevalence-adjusted bias-adjusted kappa (PABAK) may provide supplementary insights.
Standard Error and Confidence Intervals
The precision of kappa estimates is quantified through standard errors (SE) and confidence intervals. The standard error of kappa measures how much the estimated kappa would vary across different samples [91]. Smaller standard errors indicate more precise estimates, while larger standard errors suggest greater uncertainty [91].
Confidence intervals for kappa are typically calculated as: κ ± Z₁₋α/₂ × SEκ where Z₁₋α/₂ is 1.96 for 95% confidence [88]. Reporting confidence intervals helps contextualize kappa estimates, as a kappa of 0.70 with a wide confidence interval (e.g., 0.50-0.90) provides less certainty about the true agreement than the same kappa with a narrow interval (e.g., 0.65-0.75) [88] [89].
Statistical significance testing (p-values) for kappa evaluates whether the observed agreement exceeds chance levels [89]. However, with sufficient sample size, even trivial agreements beyond chance may achieve statistical significance, so effect size interpretation (kappa magnitude) is generally more informative than statistical significance alone.
Essential Reagents and Research Solutions
Research Reagent Solutions for Allergen Detection Studies
Table 5: Essential Materials for ELISA vs. PCR Comparison Studies
| Reagent/Equipment | Function in Method Comparison | Specific Requirements |
|---|---|---|
| Certified Reference Materials | Quantification standards | Matrix-matched with target food |
| Monoclonal Antibodies (ELISA) | Allergen capture and detection | Target epitope-specific, validated |
| Primers and Probes (PCR) | DNA amplification and detection | Allergen gene-specific, optimized |
| Protein Extraction Buffer | Allergen protein recovery | Compatible with food matrix |
| DNA Extraction Kit | Genomic DNA isolation | Efficient for processed foods |
| Blocking Agents | Reduce non-specific binding (ELISA) | Protein-based (BSA, non-fat milk) |
| DNA Polymerase | DNA amplification (PCR) | Thermostable, high fidelity |
| Chromogenic Substrates | Signal generation (ELISA) | Sensitive, low background |
| Thermal Cycler | DNA amplification (PCR) | Precise temperature control |
| Microplate Reader | Absorbance measurement (ELISA) | Accurate at appropriate wavelengths |
Successful comparison of ELISA and PCR methods requires careful selection of reagents validated for the specific food matrices and allergens under investigation. Certified reference materials with known allergen concentrations are particularly crucial for establishing classification thresholds and ensuring both methods are evaluating the same underlying phenomenon.
Kappa statistics provide a robust, chance-corrected framework for evaluating agreement between analytical methods like ELISA and PCR for allergen detection. While kappa has limitations, including sensitivity to prevalence and category distribution, it remains a valuable tool for method validation studies when applied and interpreted appropriately. For allergen detection in processed foods, where reliable classification directly impacts consumer safety, kappa values exceeding 0.70 represent substantial agreement, though context-specific considerations may warrant higher thresholds. By implementing proper experimental designs, selecting appropriate kappa variants, and reporting comprehensive results including confidence intervals, researchers can make informed decisions about method reliability and interchangeability in food safety testing.
For researchers and scientists in food safety and diagnostic development, the selection between Enzyme-Linked Immunosorbent Assay (ELISA) and Polymerase Chain Reaction (PCR) for allergen detection extends beyond mere technical preference. It represents a critical decision point that intersects with regulatory compliance, method validation requirements, and analytical performance in complex food matrices. With undeclared allergens consistently ranking as the leading cause of food recalls—accounting for approximately 34.1% of U.S. food recalls—the implementation of rigorously validated detection frameworks becomes paramount for both public health and regulatory adherence [19]. The global food allergen testing market reflects this heightened emphasis on reliability, projected to grow from USD 900.1 million in 2024 to USD 1,909.3 million by 2034 [19]. This article provides a comprehensive comparative analysis of ELISA and PCR validation frameworks, supported by experimental data and structured to guide method certification decisions in processed food analysis.
Comparative Method Performance: ELISA vs. PCR
Fundamental Detection Principles
ELISA and PCR methodologies operate on fundamentally different analytical principles, targeting distinct molecular markers of allergenic presence:
ELISA (Protein-Based Detection): This immunoassay technique utilizes antibody-antigen interactions to directly detect allergenic proteins. The sandwich ELISA format, which employs two antibodies binding to different epitopes on the target protein, dominates allergen testing due to its high specificity and reliable quantification capabilities [4]. The method directly measures the causative agents of allergic reactions—proteins—and is particularly effective for native and minimally processed proteins where epitope structures remain intact.
PCR (DNA-Based Detection): This molecular technique amplifies species-specific DNA sequences to indirectly indicate the presence of allergenic ingredients. By targeting genetic markers unique to allergenic sources (e.g., peanut, soy, or shellfish DNA), PCR detects the genetic blueprint rather than the allergenic protein itself [4] [5]. This approach proves particularly valuable when proteins have been denatured or altered during processing but DNA sequences remain amplifiable.
Quantitative Performance Comparison
Experimental studies directly comparing both methodologies across various food matrices provide critical performance data for validation frameworks. The table below summarizes key quantitative findings from comparative studies:
Table 1: Analytical Performance of ELISA vs. PCR Methods
| Performance Metric | ELISA | PCR | Experimental Context |
|---|---|---|---|
| Detection Limit (Beef) | 1.00% (w/w) | 0.50% (w/w) | Binary meat mixtures [8] |
| Detection Limit (Pork) | 10.0% (w/w) | 0.10% (w/w) | Binary meat mixtures [8] |
| Dynamic Range | 200-4000 mg/kg | 0.1-106 mg/kg | Crustacean shellfish detection [2] |
| Lupine Detection | 10 ppm (LOD) | Less sensitive than sandwich ELISA | Model foods (bread, biscuits, noodles) [93] |
| Matrix Interference | Significant in complex matrices | Minimal interference | Crustacean detection in fish sauce [2] |
| Quantitative Capability | Fully quantitative | Qualitative or semi-quantitative | Commercial testing applications [4] [5] |
Additional studies focusing on crustacean allergen detection further corroborate these performance trends, showing PCR's broader dynamic range and superior resistance to matrix effects compared to ELISA [2]. The fundamental difference in target molecule stability—DNA versus protein—largely explains these performance variations, particularly in processed foods where protein denaturation can compromise ELISA detection while DNA fragments remain amplifiable.
Methodological Workflows
The experimental workflows for both techniques involve distinct processes, reagents, and equipment requirements:
Figure 1: Comparative Methodological Workflows for ELISA and PCR Detection
Validation Frameworks and Regulatory Compliance
Method Validation Requirements
Validation frameworks for allergen detection methods must demonstrate reliability across multiple performance parameters, with specific emphasis on method suitability for intended applications and matrices. ISO 17025 accreditation represents the benchmark for testing laboratories, requiring demonstrated method validation across critical parameters [4]. Key validation components include:
Specificity and Cross-Reactivity Assessment: Antibody specificity in ELISA must be validated against related species and common food components to minimize false positives. Similarly, PCR primer specificity must be verified against non-target DNA sequences to ensure exclusive amplification of target allergen sequences [3] [93].
Sensitivity and Detection Limits: Method validation must establish Limit of Detection (LOD) and Limit of Quantification (LOQ) appropriate for regulatory thresholds. For example, the Codex Alimentarius specifies 20 mg/kg gluten as the threshold for "gluten-free" claims, with ELISA serving as the official testing method [3].
Matrix Effects and Robustness: Comprehensive validation requires testing across diverse product matrices (e.g., chocolate, baked goods, sauces) to identify potential interference compounds that may affect either protein extraction (ELISA) or DNA amplification (PCR) [8] [4].
Repeatability and Reproducibility: Inter-laboratory studies and statistical analysis of duplicate samples measure method precision, with PCR demonstrating 96.7% agreement versus 95.6% for ELISA in meat species identification [8].
Regulatory Recognition and Method Certification
Global regulatory frameworks exhibit varying acceptance of ELISA and PCR methods, influencing certification strategies:
Codex Alimentarius: Officially adopts ELISA as the reference method for gluten allergen testing, establishing specific thresholds for gluten-free claims [3].
United States (FDA): Recognizes both methodologies, with ELISA remaining predominant for routine allergen testing due to its direct measurement of allergenic proteins and established validation protocols [19].
European Union: Maintains specific method requirements under Regulation 1169/2011, with technical guidance provided in SANTE/11312/2021 [4].
Japan: Officially recognizes both ELISA and PCR as testing methods, establishing a food allergen threshold of 10 μg/g [3].
Certification processes require demonstration of method performance through interlaboratory studies, reference material analysis, and proficiency testing. Laboratories must maintain detailed validation records including calibration curves, extraction efficiency data, and statistical analysis of repeatability measures.
Experimental Protocols and Research Reagents
Key Research Reagent Solutions
Table 2: Essential Research Reagents for Allergen Detection Methods
| Reagent Category | Specific Examples | Function & Importance | Method Application |
|---|---|---|---|
| Capture/Detection Antibodies | Anti-Ara h 1 (peanut); Anti-Gal d 1 (egg) | Specifically bind target allergenic proteins; determine assay specificity | ELISA |
| Species-Specific Primers/Probes | 12S mitochondrial gene targets (shrimp, crab); Tropomyosin gene | Amplify unique DNA sequences of allergenic species; determine assay specificity | PCR |
| Protein Extraction Buffers | PBS with Tween 20; commercial extraction kits | Efficiently extract proteins from complex food matrices; critical for quantification | ELISA |
| DNA Extraction/Purification Kits | DNG-PLUS; commercial DNA isolation kits | Isolate amplifiable DNA while removing PCR inhibitors | PCR |
| Enzyme-Substrate Systems | Horseradish peroxidase with OPD/TMB | Generate measurable signal proportional to allergen concentration | ELISA |
| DNA Amplification Master Mixes | TaqMan with MGB probes; SYBR Green | Enable specific DNA amplification with real-time detection | PCR |
| Reference Materials | Certified allergenic ingredients; in-house controls | Validate method accuracy and establish calibration curves | ELISA & PCR |
Standardized Experimental Protocols
For method validation studies, standardized protocols ensure comparable results across laboratories:
Sandwich ELISA Protocol for Protein Allergens [4] [93]:
- Coating: Microplate wells are coated with capture antibodies specific to target allergen (e.g., anti-tropomyosin for shellfish) in carbonate-bicarbonate buffer (pH 9.6), incubated overnight at 4°C.
- Blocking: Non-specific binding sites are blocked with 3% skim milk or BSA in PBST for 2 hours at room temperature.
- Sample Incubation: Protein extracts from test samples are added and incubated for 30-60 minutes, allowing allergen binding to capture antibodies.
- Detection Antibody: Enzyme-conjugated detection antibody is added, forming the "sandwich" complex, incubated for 30-60 minutes.
- Signal Development: Enzyme substrate (e.g., OPD, TMB) is added, producing colorimetric change measurable at specific wavelengths.
- Quantification: Optical density is measured spectrophotometrically, with allergen concentration calculated against standard curves.
Real-Time PCR Protocol for Allergen DNA Detection [2] [8]:
- DNA Extraction: Samples undergo mechanical and chemical lysis, followed by DNA purification using silica columns or magnetic beads to remove PCR inhibitors.
- Primer/Probe Design: Species-specific primers target conserved genes (e.g., mitochondrial 12S for crustaceans) with TaqMan MGB probes enhancing specificity.
- Amplification Reaction: PCR mix contains template DNA, primers, probes, dNTPs, and DNA polymerase in optimized buffer.
- Thermal Cycling: Typically 40-45 cycles of denaturation (95°C), annealing (50-60°C), and extension (72°C) in real-time thermocycler.
- Fluorescence Detection: Probe cleavage releases reporter dye fluorescence measured during each cycle.
- Data Analysis: Threshold cycle (Ct) values determine presence/absence; quantification against standard curves for semi-quantitative analysis.
Integrated Decision Framework
Selection between ELISA and PCR methodologies requires systematic consideration of multiple analytical and regulatory factors:
Figure 2: Method Selection Decision Framework for Allergen Detection
Complementary Applications and Future Perspectives
Rather than mutually exclusive alternatives, ELISA and PCR frequently serve complementary roles in comprehensive allergen control plans. Integrated approaches leveraging both methodologies provide superior detection coverage across diverse product types and processing conditions [5]. Industry trends indicate growing adoption of confirmatory testing strategies where positive ELISA screens undergo PCR verification, particularly for high-risk products or disputed results [19].
Emerging technologies including mass spectrometry and biosensors present future alternatives, with LC-MS/MS methods achieving detection limits as low as 0.01 ng/mL for specific allergenic proteins through targeted peptide analysis [26] [3]. However, these techniques remain predominantly supplemental rather than replacement technologies for regulatory testing purposes.
For researchers and drug development professionals, the validation framework selection fundamentally hinges on the specific analytical question: ELISA provides direct measurement of the causative agent (protein) and remains the regulatory preferred method for quantitative analysis, while PCR offers superior sensitivity and processing resistance for qualitative detection of allergenic ingredients. The evolving regulatory landscape and technological advances continue to shape method certification requirements, emphasizing the necessity for ongoing method validation and proficiency demonstration in accredited laboratory settings.
Conclusion
The choice between ELISA and PCR is not a matter of superiority but of strategic application, dictated by the specific food matrix, the degree of processing, and the required output. ELISA remains the gold standard for direct, quantitative protein detection, particularly for allergens like gluten and milk. In contrast, PCR offers a powerful, sensitive alternative for detecting allergen sources in highly processed foods where proteins may be denatured. An integrated approach, leveraging the complementary strengths of both methods, often provides the most comprehensive risk assessment. Future directions point towards the adoption of biosensors, mass spectrometry, and AI-driven predictive analytics, which promise to deliver faster, more sensitive, and non-destructive allergen detection, further revolutionizing food safety protocols and clinical research.
References
- 1. ELISA Assay Technique | Thermo Fisher Scientific - AR [https://www.thermofisher.com/ar/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 2. Comparison of real-time PCR and ELISA for the detection ... [https://pubmed.ncbi.nlm.nih.gov/33617420/]
- 3. Research progress on detection methods for food allergens [https://www.sciencedirect.com/science/article/abs/pii/S0889157524009402]
- 4. PCR & ELISA Allergen Testing: How Manufacturers Detect ... [https://certified-laboratories.com/blog/pcr-elisa-allergen-testing-how-manufacturers-detect-hidden-risks/]
- 5. Food allergen testing: should you use ELISA, PCR, or LFA ... [https://blog.invitek.com/articles/foodsafetyintegrity/food-allergen-testing-should-you-use-elisa-pcr-or-lfa-methods]
- 6. Enzyme Linked Immunosorbent Assay - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK555922/]
- 7. The principle and method of ELISA [https://www.mblbio.com/bio/g/support/method/elisa.html]
- 8. Comparison of real-time PCR and ELISA-based methods ... [https://www.sciencedirect.com/science/article/abs/pii/S0956713516303802]
- 9. Polymerase Chain Reaction (PCR) - StatPearls - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK589663/]
- 10. Polymerase Chain Reaction (PCR) Fact Sheet [https://www.genome.gov/about-genomics/fact-sheets/Polymerase-Chain-Reaction-Fact-Sheet]
- 11. PCR: The Technology That Launched Biotechnology [https://www.nsf.gov/impacts/polymerase-chain-reaction]
- 12. PCR Amplification | An Introduction to PCR Methods [https://www.promega.com/resources/guides/nucleic-acid-analysis/pcr-amplification/]
- 13. The Science of Allergen Detection: Lateral Flow and ELISA ... [https://www.prognosis-biotech.com/news-events/the-science-of-allergen-detection-lateral-flow-and-elisa-explained/]
- 14. Development and Validation of ELISA for In Vitro Diagnosis ... [https://www.mdpi.com/2673-8112/5/7/108]
- 15. Detection of Allergenic Proteins in Foodstuffs: Advantages ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8949930/]
- 16. Development of PCR Methods for Detecting Wheat and Maize ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12551070/]
- 17. Development of a real-time PCR and a sandwich ELISA for ... [https://pubmed.ncbi.nlm.nih.gov/15186093/]
- 18. Improved Allergen Detection in Processed Food [https://www.eurofins.de/food-analysis/food-news/food-testing-news/improved-allergen-detection-in-processed-food/]
- 19. The Growing Importance of Allergen Testing: How Certified ... [https://fsns.com/the-growing-importance-of-allergen-testing-how-certified-group-ensures-food-safety-with-elisa/]
- 20. Methods to determine limit of detection and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5496743/]
- 21. Sensibility and Specificity of the VitaPCR™ SARS-CoV-2 ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9866422/]
- 22. The difference between sensitivity and detection range in ... [https://www.bosterbio.com/blog/post/the-difference-between-sensitivity-and-the-minimum-level-of-assay-range-in-elisa-testing?srsltid=AfmBOoo7SlGxxmifIcm9puSuFz-OtJvGpL1auhDaXHCsLu1-evFRDeL9]
- 23. Review of allergen analytical testing methodologies [https://www.food.gov.uk/research/review-of-allergen-analytical-testing-methodologies-allergen-testing-workflows-to-support-incident-management]
- 24. What Is the Sensitivity Limit of ELISA Assays? [https://synapse.patsnap.com/article/what-is-the-sensitivity-limit-of-elisa-assays]
- 25. A Comparative Study of Real-Time RT-PCR–Based SARS ... [https://www.sciencedirect.com/science/article/pii/S1525157821001185]
- 26. Innovations shaping the future of food allergen testing [https://allergenbureau.net/innovations-shaping-the-future-of-food-allergen-testing/]
- 27. A rapid and highly sensitive biomarker detection platform ... [https://www.nature.com/articles/s41598-020-75011-x]
- 28. Food Allergen Testing Market [https://www.futuremarketinsights.com/reports/food-allergen-testing-market]
- 29. Allergy Diagnostics Global Overview Report 2025 | Market ... [https://finance.yahoo.com/news/allergy-diagnostics-global-overview-report-104100046.html]
- 30. Food Allergies [https://www.fda.gov/food/nutrition-food-labeling-and-critical-foods/food-allergies]
- 31. A digital ELISA for multiplexed detection of allergen‐ ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12617554/]
- 32. ELISA Assay Validation: Best Practices, Methods, and Key ... [https://www.nebiolab.com/elisa-assay-validation-guide/]
- 33. A comparison of PCR and ELISA methods to detect different ... [https://parasitesandvectors.biomedcentral.com/articles/10.1186/s13071-021-04976-z]
- 34. ELISA Data Analysis Instructions [https://www.bosterbio.com/elisa-data-analysis-instructions?srsltid=AfmBOoqJl1yGiqYti16UnGmdYjXRUNi2OecvTte9BidIyNWHKwNV4-5Y]
- 35. A reliable enzyme-linked immunosorbent assay for the ... [https://www.sciencedirect.com/science/article/pii/S2590157524001184]
- 36. ELISA Kit Validation and Quality Testing [https://www.thermofisher.com/us/en/home/life-science/antibodies/immunoassays/elisa-kits/elisa-kit-validation-quality-testing.html]
- 37. A novel ELISA test to detect soy in highly processed foods [https://www.sciencedirect.com/science/article/pii/S0889157521005032]
- 38. Advances in Food Processing Techniques for Allergenicity ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12651889/]
- 39. Real-time PCR methods for identification and stability ... [https://www.frontiersin.org/journals/microbiology/articles/10.3389/fmicb.2024.1360241/full]
- 40. Improved multi-food allergen analysis of processed ... [https://pubmed.ncbi.nlm.nih.gov/39078454/]
- 41. Why allergen detection by ELISA is insufficient to protect ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3723902/]
- 42. Food Allergen Detection for Safer, Compliant Production [https://www.hygiena.com/food-safety/allergen-detection]
- 43. of Performance and ELISA methods for the determination of... PCR [https://pubmed.ncbi.nlm.nih.gov/24144233/]
- 44. A new real-time PCR quantitative approach for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0889157518302850]
- 45. An improved duplex real-time PCR method for the ... [https://www.sciencedirect.com/science/article/abs/pii/S0956713522007101]
- 46. A group-specific, quantitative real-time PCR assay for ... [https://pubmed.ncbi.nlm.nih.gov/29120774/]
- 47. Tests for the detection of gluten in food | R-Biopharm [https://food.r-biopharm.com/analytes/food-allergens/gliadin-gluten/]
- 48. Gluten Detection Methods and Their Critical Role in Assuring ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6949940/]
- 49. ELISA Assay Technique | Thermo Fisher Scientific - US [https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/overview-elisa.html]
- 50. Veratox® for Gliadin R5 - ELISA test for gluten - Generon [https://www.generon-food-safety.com/product/veratox-for-gliadin-r5-elisa-test-for-gluten/]
- 51. PCR in allergen analysis: a controversial issue - R-Biopharm [https://food.r-biopharm.com/news/pcr-in-allergen-analysis-a-controversial-issue/]
- 52. Lateral flow assays: Progress and evolution of recent ... [https://www.sciencedirect.com/science/article/pii/S2590006424002497]
- 53. What's the difference between LFD , ELISA and PCR testing? [https://emportllc.com/lfd-elisa-pcr-testing/]
- 54. Lateral flow assays - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4986465/]
- 55. A comprehensive review of competitive lateral flow assays ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d4lc01075b]
- 56. Advantages and Limitations of LFAs - Lateral Flow [https://hiyka.com/lateral-flow/advantages-and-limitations-of-lfas/?srsltid=AfmBOoqiP3VYqyYH4vVALGWNDnTLy5IMJQi7i6M-OyTIh-F1iER4m8M3]
- 57. Comparison of commercial lateral flow immunoassays and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7323682/]
- 58. Comparative evaluation of in-house ELISA and two ... [https://www.nature.com/articles/s41598-025-97050-y]
- 59. Lateral Flow Assay: A Summary of Recent Progress for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10526804/]
- 60. What Are the Differences Between ELISA and PCR for ... [https://synapse.patsnap.com/article/what-are-the-differences-between-elisa-and-pcr-for-diagnostic-testing]
- 61. What's ELISA? Is it different than PCR when testing COVID [https://www.rocker.com.tw/en/application/elisa/]
- 62. A reliable enzyme-linked immunosorbent assay for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10900753/]
- 63. Detection of Food Allergens by Taqman Real-Time PCR ... [https://pubmed.ncbi.nlm.nih.gov/28315214/]
- 64. Determining Matrix Effects in Complex Food Samples [https://www.waters.com/blog/understanding-sample-complexity-determining-matrix-effects-in-complex-food-samples/?srsltid=AfmBOoqZg6nCvhnLNhJnSUi02Q5MlRPhDNtfkSdeKSfppfQjiVb27zg8]
- 65. Addressing Complex Matrix Interference Improves ... [https://pubmed.ncbi.nlm.nih.gov/31339301/]
- 66. Strategies for the Detection and Elimination of Matrix ... [https://www.chromatographyonline.com/view/strategies-detection-and-elimination-matrix-effects-quantitative-lc-ms-analysis-0]
- 67. Enhanced Matrix Removal - Lipid - Recommended Protocols [https://www.agilent.com/en-us/products/sample-preparation/sample-preparation-methods/quechers/enhanced-matrix-removal-lipid-recommended-protocols?srsltid=AfmBOorotk7MM8OgllQ_zRo_VGhQONnMcZiOXuPKpbaUZo40hHRuvYER]
- 68. Matrix removal in state of the art sample preparation ... [https://www.sciencedirect.com/science/article/abs/pii/S000326701630232X]
- 69. Why are Sensitivity and Specificity Important Parameters for... - Enzo [https://www.enzo.com/note/why-are-sensitivity-and-specificity-important-parameters-for-an-elisa/]
- 70. Performance Evaluation of a Commercial Real-Time PCR ... [https://www.mdpi.com/2304-8158/13/4/609]
- 71. tests ELISA , Performance , Recovery, linearity Specificity Sensitivity [https://diagnopal.ca/elisa-performance-tests-specificity-sensitivity-recovery-linearity/]
- 72. Collaborative trial validation of a new multiplex real-time ... [https://link.springer.com/article/10.1007/s00003-022-01385-x]
- 73. ELISA Sensitivity and Specificity [https://www.mybiosource.com/learn/elisa-sensitivity-and-specificity/]
- 74. Application of PCR-ELISA in Molecular Diagnosis - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4058250/]
- 75. Sensitivity, Specificity, Positive Predictive Value, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8156826/]
- 76. An overview of ELISA: a review and update on best laboratory ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11808753/]
- 77. Comparative Evaluation of RUT, PCR and ELISA Tests for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4961986/]
- 78. Sensitivity and Specificity of ELISA Tests Detecting BP180 ... [https://pubmed.ncbi.nlm.nih.gov/41222109/]
- 79. High-Throughput Data Analysis for qPCR [https://www.neb.com/en-us/tools-and-resources/feature-articles/development-of-a-high-throughput-data-analysis-method-for-quantitative-real-time-pcr?srsltid=AfmBOopvhTcir_V5RHpRKIfd8MiFJhYVecBuRuCpHbE25RWEdcAbL-VR]
- 80. How to interpret the dynamic range of an ELISA [https://www.abcam.com/en-us/technical-resources/guides/elisa-guide/elisa-dynamic-range]
- 81. Comparison of Real-Time PCR, Conventional ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC179803/]
- 82. ELISA Sensitivity and Specificity [https://www.mybiosource.com/learn/elisa-sensitivity-and-specificity-2/]
- 83. Economic Comparison of Serologic and Molecular ... [https://pubmed.ncbi.nlm.nih.gov/30302060/]
- 84. Economic Comparison of Serologic and Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6170887/]
- 85. Interrater reliability: the kappa statistic - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3900052/]
- 86. Relationship Between Intraclass Correlation (ICC ... - IRRsim [https://irrsim.bryer.org/articles/IRRsim.html]
- 87. Cohen's Kappa Statistic: Definition & Example [https://www.statology.org/cohens-kappa-statistic/]
- 88. Cohen's kappa [https://en.wikipedia.org/wiki/Cohen%27s_kappa]
- 89. Kappa statistics for Attribute Agreement Analysis - Minitab [https://support.minitab.com/en-us/minitab/help-and-how-to/quality-and-process-improvement/measurement-system-analysis/how-to/attribute-agreement-analysis/attribute-agreement-analysis/interpret-the-results/all-statistics-and-graphs/kappa-statistics/]
- 90. Kappa statistic considerations in evaluating inter-rater ... [https://bmccancer.biomedcentral.com/articles/10.1186/s12885-023-11325-z]
- 91. Cohen's Kappa: Measuring Inter-Rater Agreement [https://numiqo.com/tutorial/cohens-kappa]
- 92. A Comparison of Reliability Coefficients for Ordinal Rating ... [https://link.springer.com/article/10.1007/s00357-021-09386-5]
- 93. Validation and comparison of a sandwich ELISA, two ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814613002549]